

C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19
- PMID: 34852172
- PMCID: PMC8945302
- DOI: 10.1182/bloodadvances.2021005246
C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19
Erratum in
-
Aiello S, Gastoldi S, Galbusera M, et al. C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19. Blood Adv. 2022;6(3):866-881.Blood Adv. 2022 May 10;6(9):2812. doi: 10.1182/bloodadvances.2022007722. Blood Adv. 2022. PMID: 35507372 Free PMC article. No abstract available.
Abstract
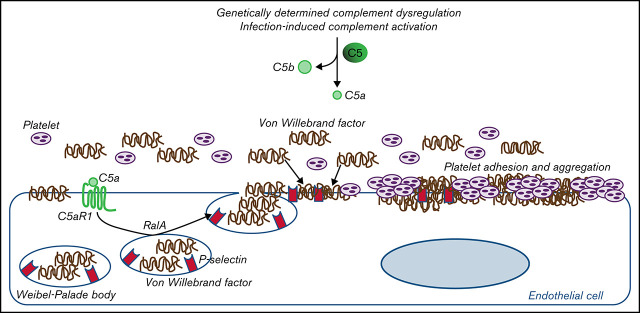
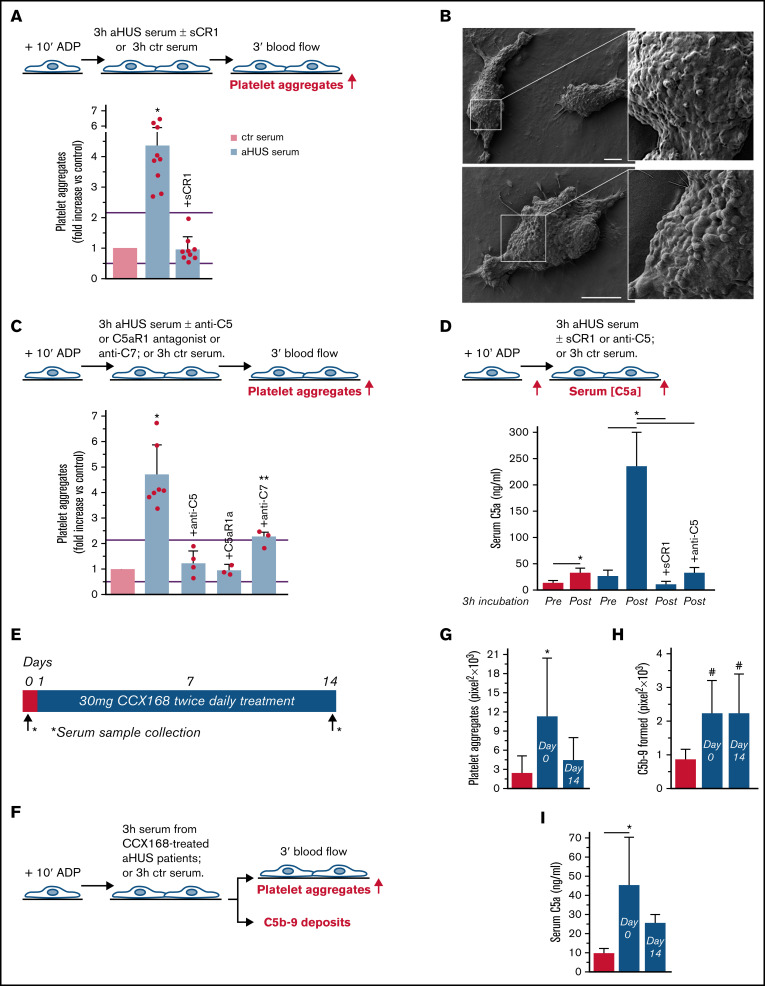
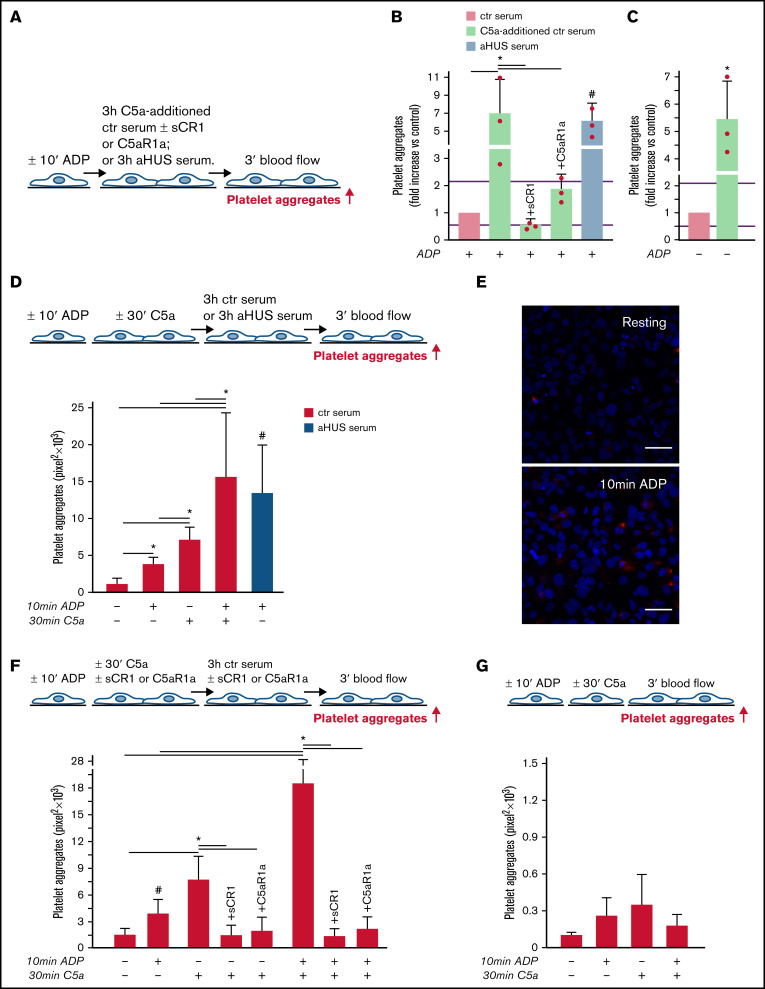
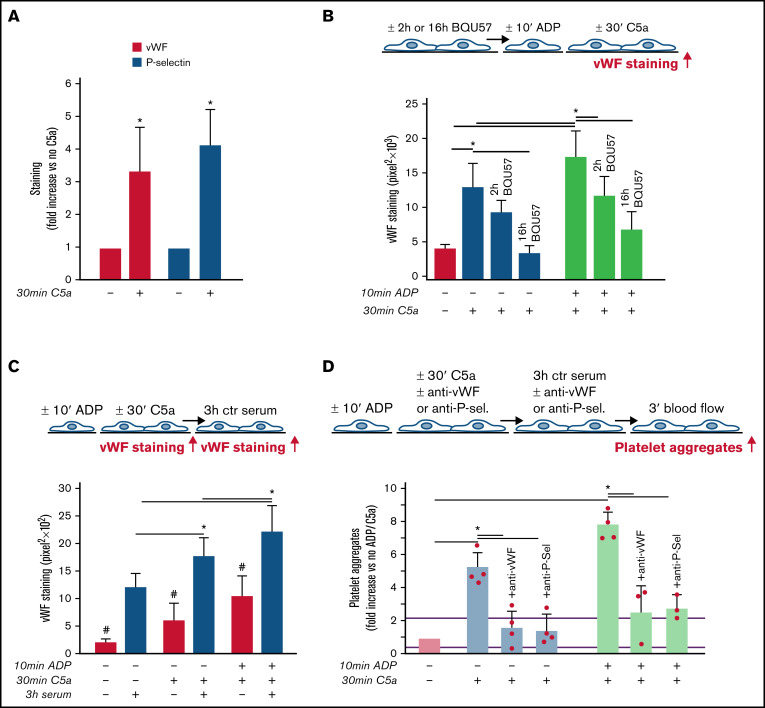
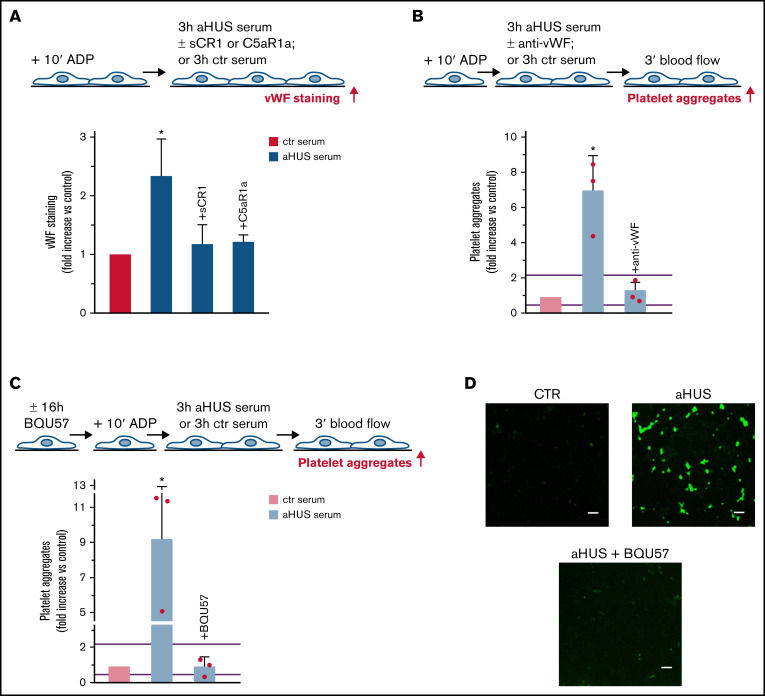
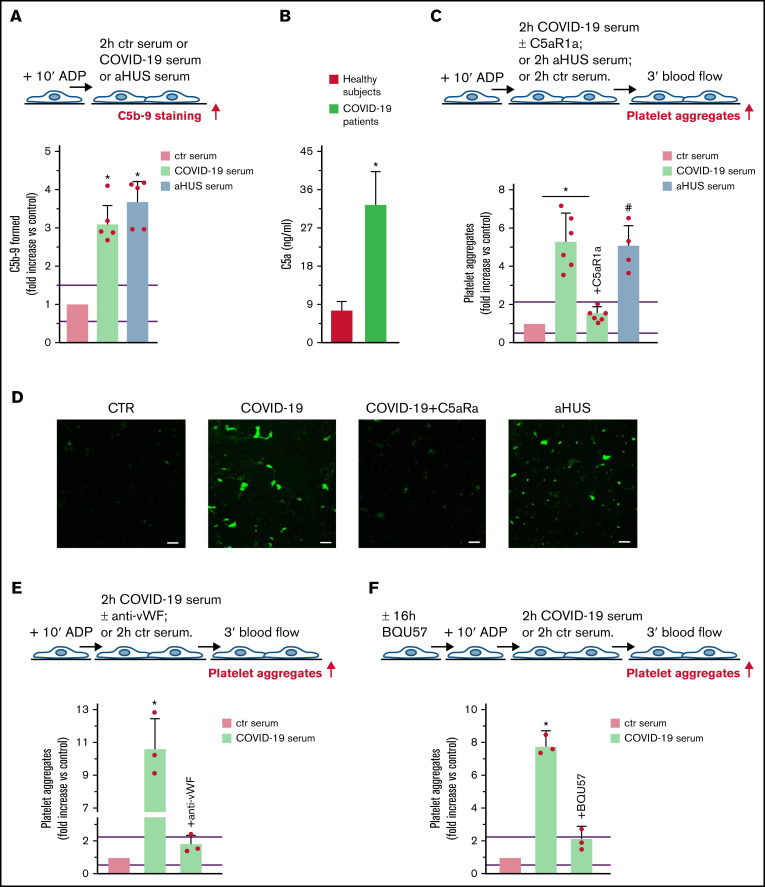
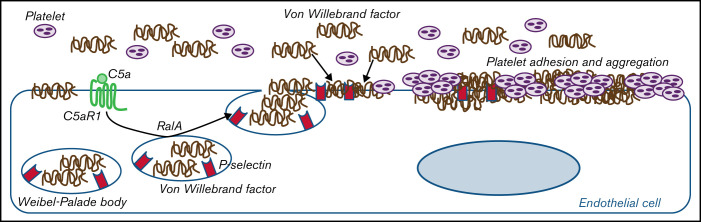
Unrestrained activation of the complement system till the terminal products, C5a and C5b-9, plays a pathogenetic role in acute and chronic inflammatory diseases. In endothelial cells, complement hyperactivation may translate into cell dysfunction, favoring thrombus formation. The aim of this study was to investigate the role of the C5a/C5aR1 axis as opposed to C5b-9 in inducing endothelial dysfunction and loss of antithrombogenic properties. In vitro and ex vivo assays with serum from patients with atypical hemolytic uremic syndrome (aHUS), a prototype rare disease of complement-mediated microvascular thrombosis due to genetically determined alternative pathway dysregulation, and cultured microvascular endothelial cells, demonstrated that the C5a/C5aR1 axis is a key player in endothelial thromboresistance loss. C5a added to normal human serum fully recapitulated the prothrombotic effects of aHUS serum. Mechanistic studies showed that C5a caused RalA-mediated exocytosis of von Willebrand factor (vWF) and P-selectin from Weibel-Palade bodies, which favored further vWF binding on the endothelium and platelet adhesion and aggregation. In patients with severe COVID-19 who suffered from acute activation of complement triggered by severe acute respiratory syndrome coronavirus 2 infection, we found the same C5a-dependent pathogenic mechanisms. These results highlight C5a/C5aR1 as a common prothrombogenic effector spanning from genetic rare diseases to viral infections, and it may have clinical implications. Selective C5a/C5aR1 blockade could have advantages over C5 inhibition because the former preserves the formation of C5b-9, which is critical for controlling bacterial infections that often develop as comorbidities in severely ill patients. The ACCESS trial registered at www.clinicaltrials.gov as #NCT02464891 accounts for the results related to aHUS patients treated with CCX168.
© 2022 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Figures
References
-
- Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V. Endothelial cells: source, barrier, and target of defensive mediators. Immunol Rev. 2016;274(1):307-329. - PubMed
-
- Lindemann S, Krämer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost. 2007;5(suppl 1):203-211. - PubMed
-
- Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058-1066. - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
