

Disease progression and clinical outcomes in telomere biology disorders
- PMID: 34852175
- PMCID: PMC8952184
- DOI: 10.1182/blood.2021013523
Disease progression and clinical outcomes in telomere biology disorders
Abstract
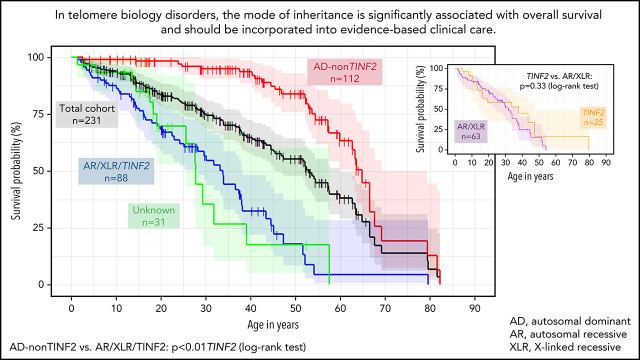
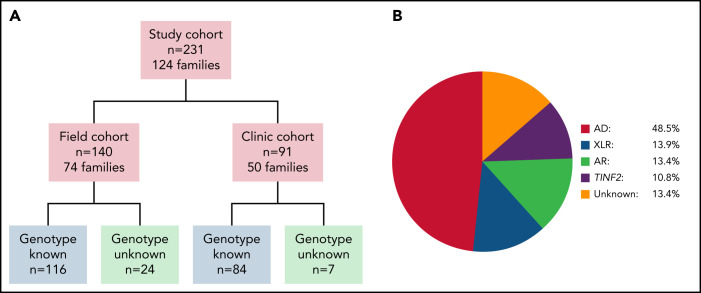
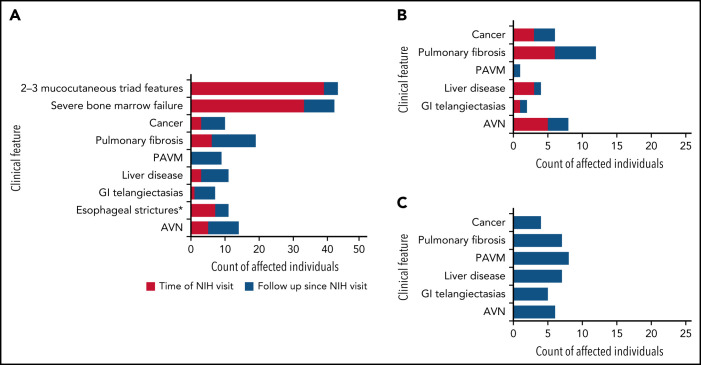
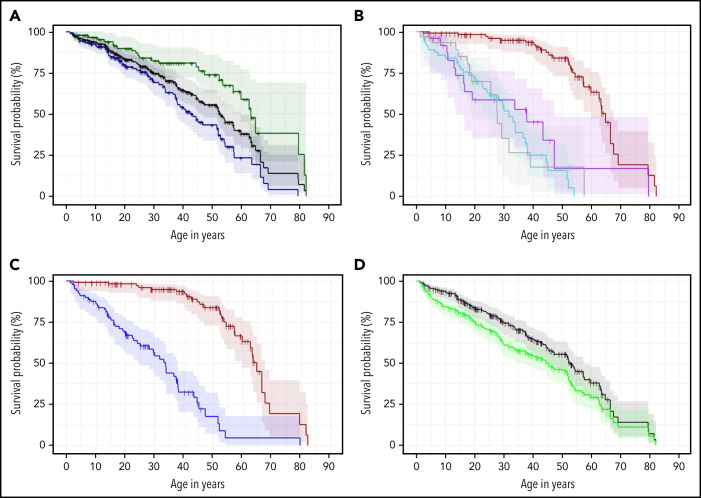
Dyskeratosis congenita related telomere biology disorders (DC/TBDs) are characterized by very short telomeres caused by germline pathogenic variants in telomere biology genes. Clinical presentations can affect all organs, and inheritance patterns include autosomal dominant (AD), autosomal recessive (AR), X-linked (XLR), or de novo. This study examined the associations between mode of inheritance with phenotypes and long-term clinical outcomes. Two hundred thirty-one individuals with DC/TBDs (144 male, 86.6% known genotype, median age at diagnosis 19.4 years [range 0 to 71.6]), enrolled in the National Cancer Institute's Inherited Bone Marrow Failure Syndrome Study, underwent detailed clinical assessments and longitudinal follow-up (median follow-up 5.2 years [range 0 to 36.7]). Patients were grouped by inheritance pattern, considering AD-nonTINF2, AR/XLR, and TINF2 variants separately. Severe bone marrow failure (BMF), severe liver disease, and gastrointestinal telangiectasias were more prevalent in AR/XLR or TINF2 disease, whereas pulmonary fibrosis developed predominantly in adults with AD disease. After adjusting for age at DC/TBD diagnosis, we observed the highest cancer risk in AR/XLR individuals. At last follow-up, 42% of patients were deceased with a median overall survival (OS) of 52.8 years (95% confidence interval [CI] 45.5-57.6), and the hematopoietic cell or solid organ transplant-free median survival was 45.3 years (95% CI 37.4-52.1). Significantly better OS was present in AD vs AR/XLR/TINF2 disease (P < .01), while patients with AR/XLR and TINF2 disease had similar survival probabilities. This long-term study of the clinical manifestations of DC/TBDs creates a foundation for incorporating the mode of inheritance into evidence-based clinical care guidelines and risk stratification in patients with DC/TBDs. This trial was registered at www.clinicaltrials.gov as #NCT00027274.
© 2022 by The American Society of Hematology.
Figures
Comment in
-
Telomere biology disorders: ends and (genetic) means.Blood. 2022 Mar 24;139(12):1776-1777. doi: 10.1182/blood.2021014855. Blood. 2022. PMID: 35323880 No abstract available.
References
-
- Zinsser F. Atrophia cutis reticularis cum pigmentatione dystrophia unguium et leukoplakia oris; poikiloderma atrophicans vascularis Jacobi. Inkonographia Dermatologia. 1906;5:219-223.
-
- Heiss NS, Knight SW, Vulliamy TJ, et al. . X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19(1):32-38. - PubMed
-
- Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402(6761):551-555. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
