

The epitope arrangement on flavivirus particles contributes to Mab C10's extraordinary neutralization breadth across Zika and dengue viruses
- PMID: 34852239
- PMCID: PMC8724787
- DOI: 10.1016/j.cell.2021.11.010
The epitope arrangement on flavivirus particles contributes to Mab C10's extraordinary neutralization breadth across Zika and dengue viruses
Abstract
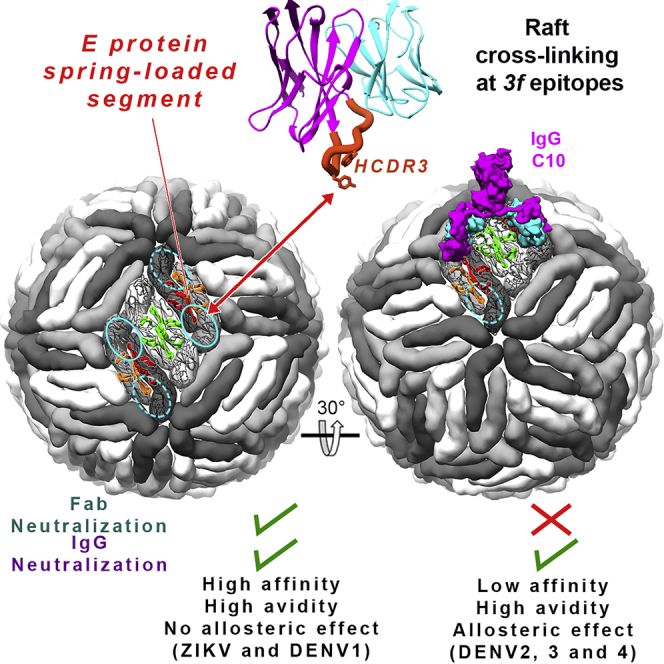
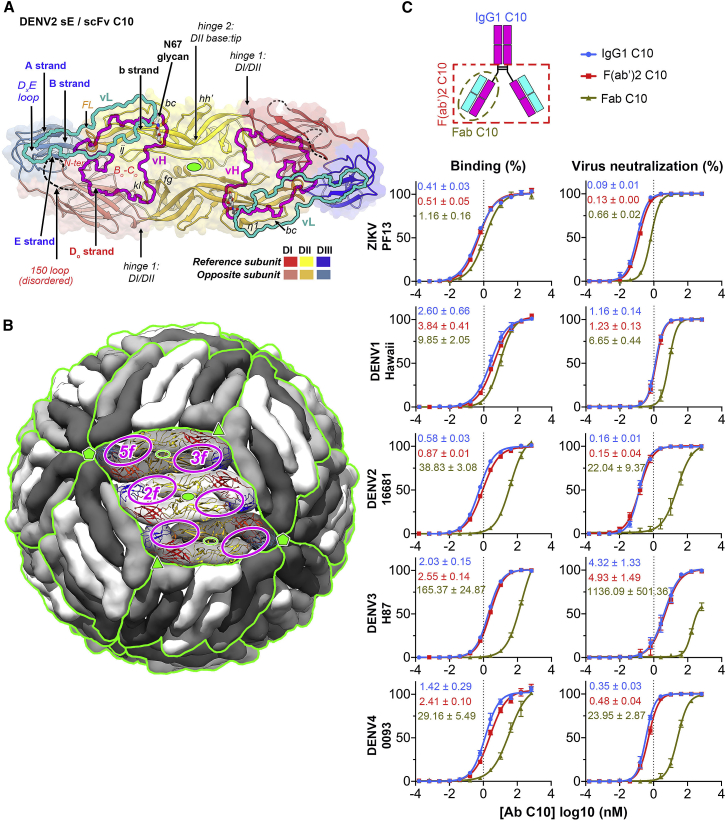
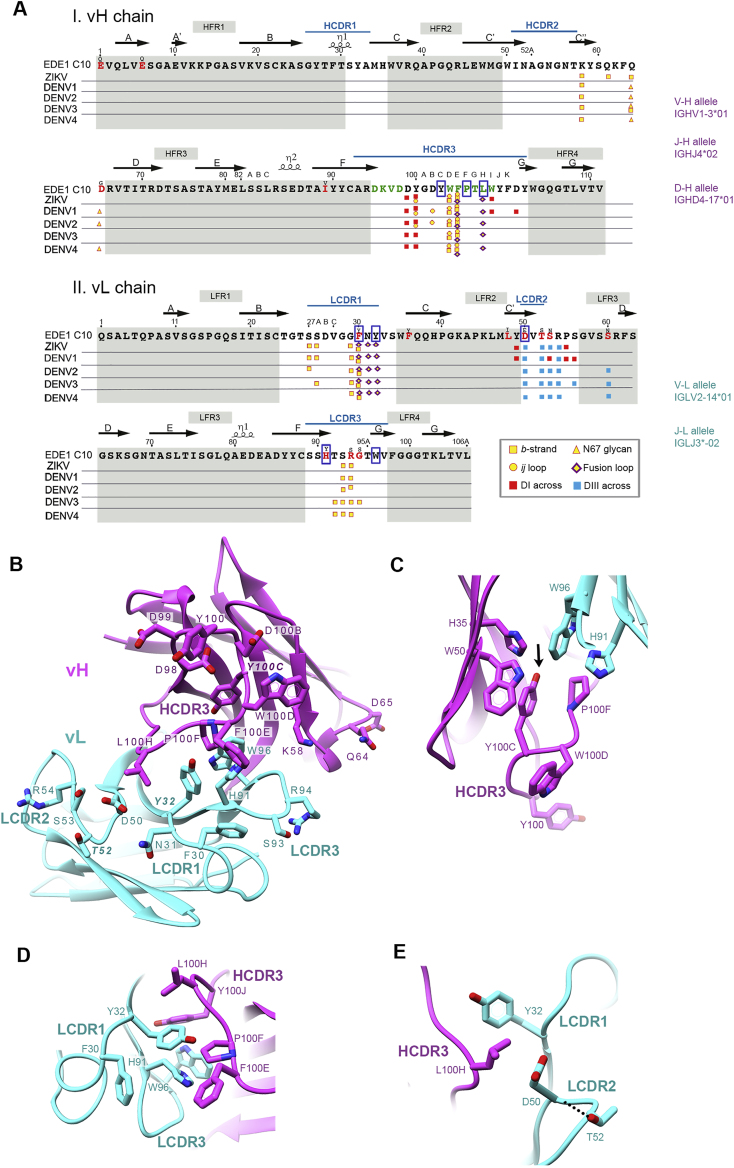
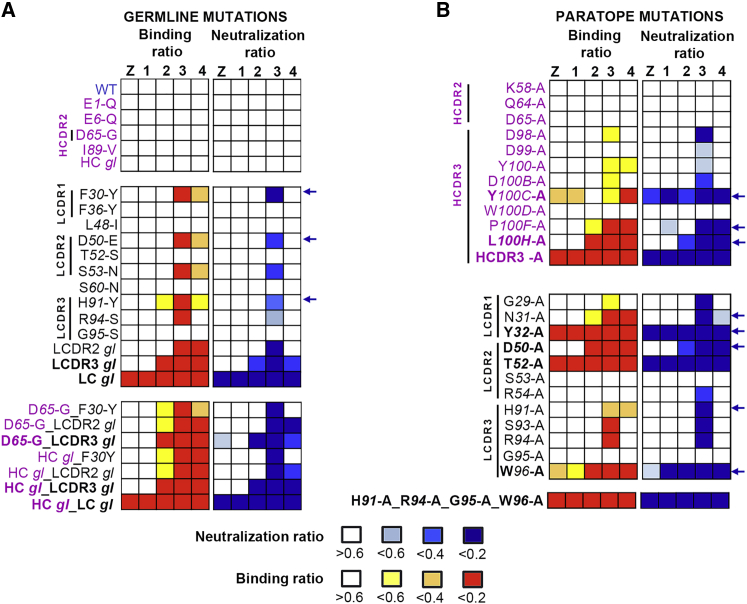
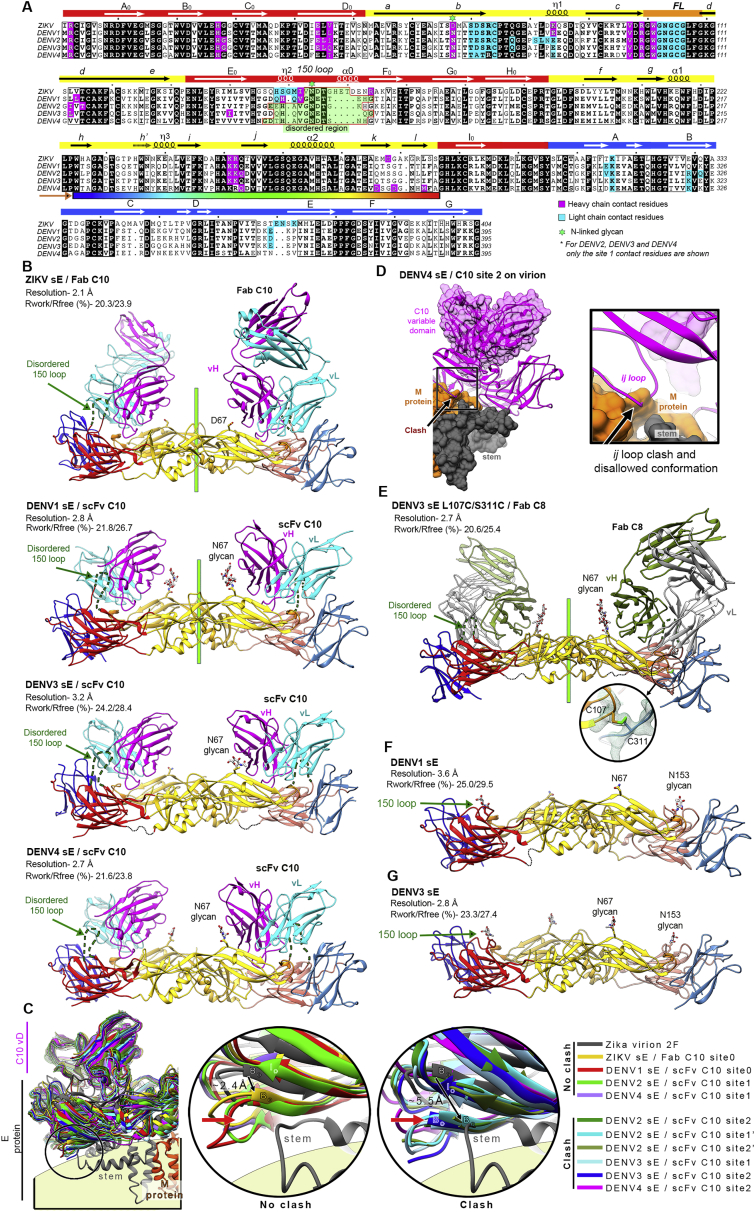
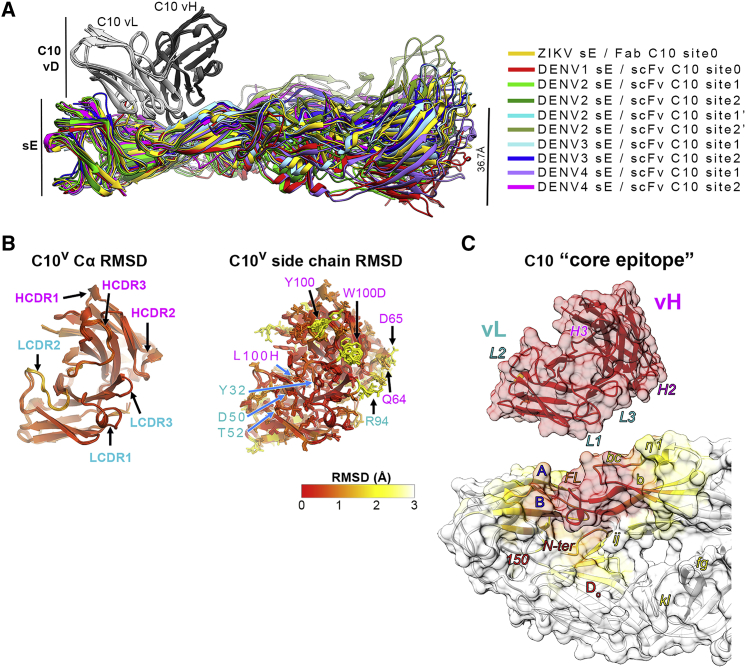
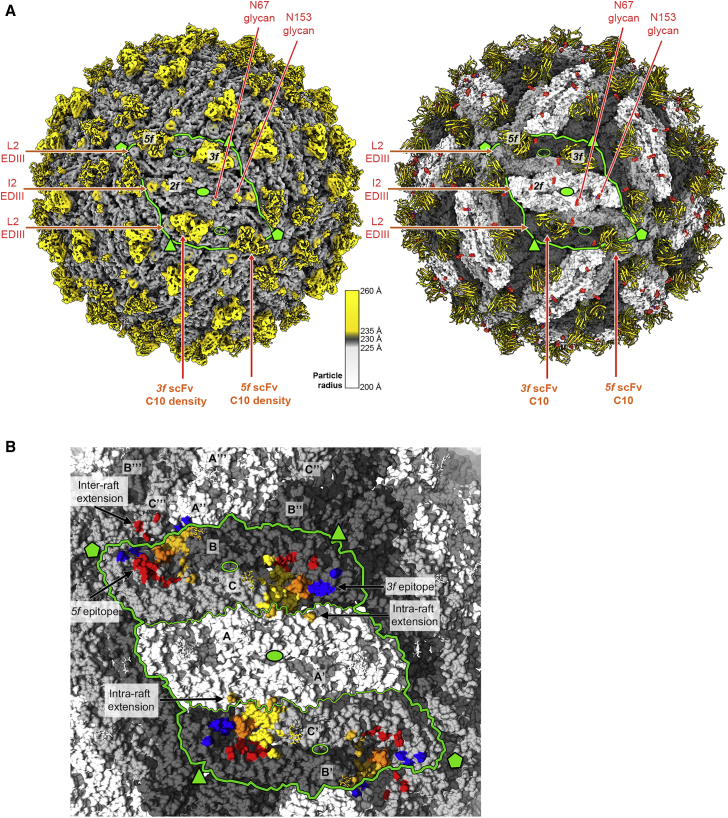
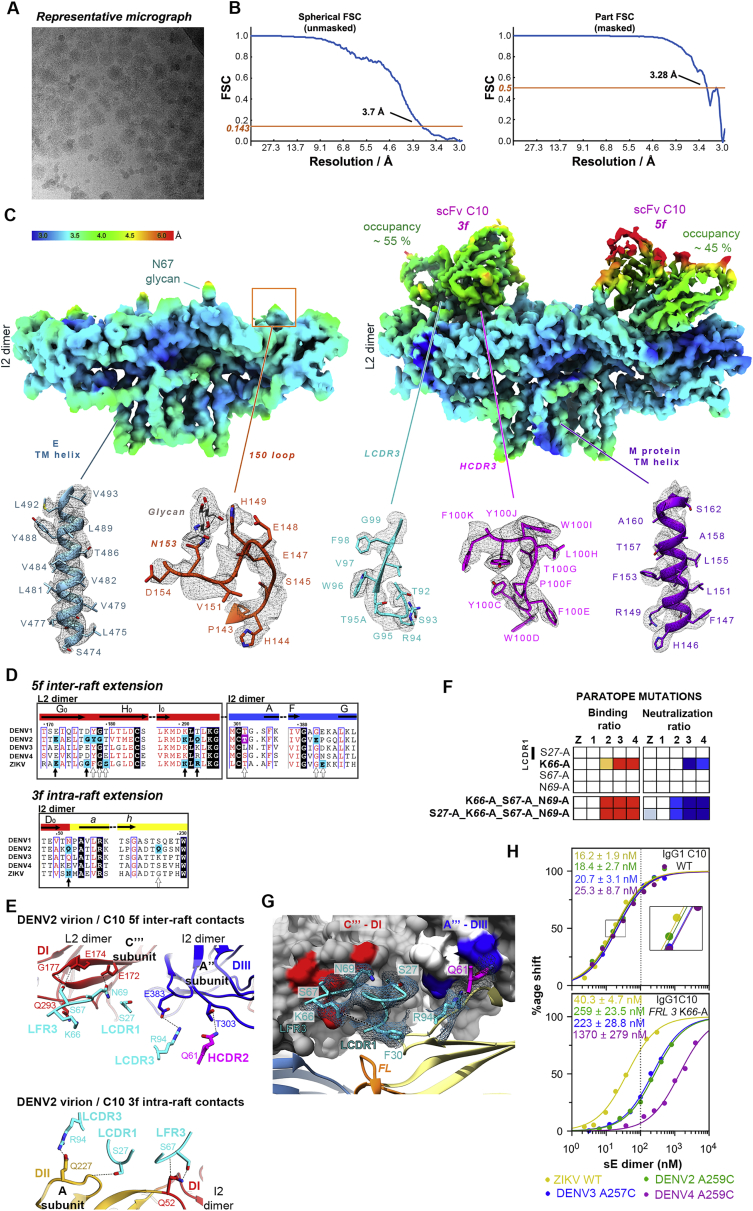
The human monoclonal antibody C10 exhibits extraordinary cross-reactivity, potently neutralizing Zika virus (ZIKV) and the four serotypes of dengue virus (DENV1-DENV4). Here we describe a comparative structure-function analysis of C10 bound to the envelope (E) protein dimers of the five viruses it neutralizes. We demonstrate that the C10 Fab has high affinity for ZIKV and DENV1 but not for DENV2, DENV3, and DENV4. We further show that the C10 interaction with the latter viruses requires an E protein conformational landscape that limits binding to only one of the three independent epitopes per virion. This limited affinity is nevertheless counterbalanced by the particle's icosahedral organization, which allows two different dimers to be reached by both Fab arms of a C10 immunoglobulin. The epitopes' geometric distribution thus confers C10 its exceptional neutralization breadth. Our results highlight the importance not only of paratope/epitope complementarity but also the topological distribution for epitope-focused vaccine design.
Keywords: Dengue virus; Flaviviruses; X-ray crystallography; Zika virus; broadly neutralizing antibodies; cryo-EM; vaccine design.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.A.R. is a board member and shareholder of EureKARE and MELETIUS Therapeutics. G.R.S. is member of the GSK Vaccines Scientific Advisory Board and a founding shareholder of RQ Biotechnology. G.R.S., F.A.R., P.G.-C., M.-C.V, A.R., S.D., and J.M. are authors in a patent concerning EDE mAbs, including C8 and C10:US20180037611A1 (2018): Anti-dengue vaccines and antibodies. G.R.S., F.A.R., M.-C.V., A.R., and J.M. are authors of patent CA3066488A1 (2017): Neutralising antibody against dengue for use in a method of prevention and/or treatment.
Figures
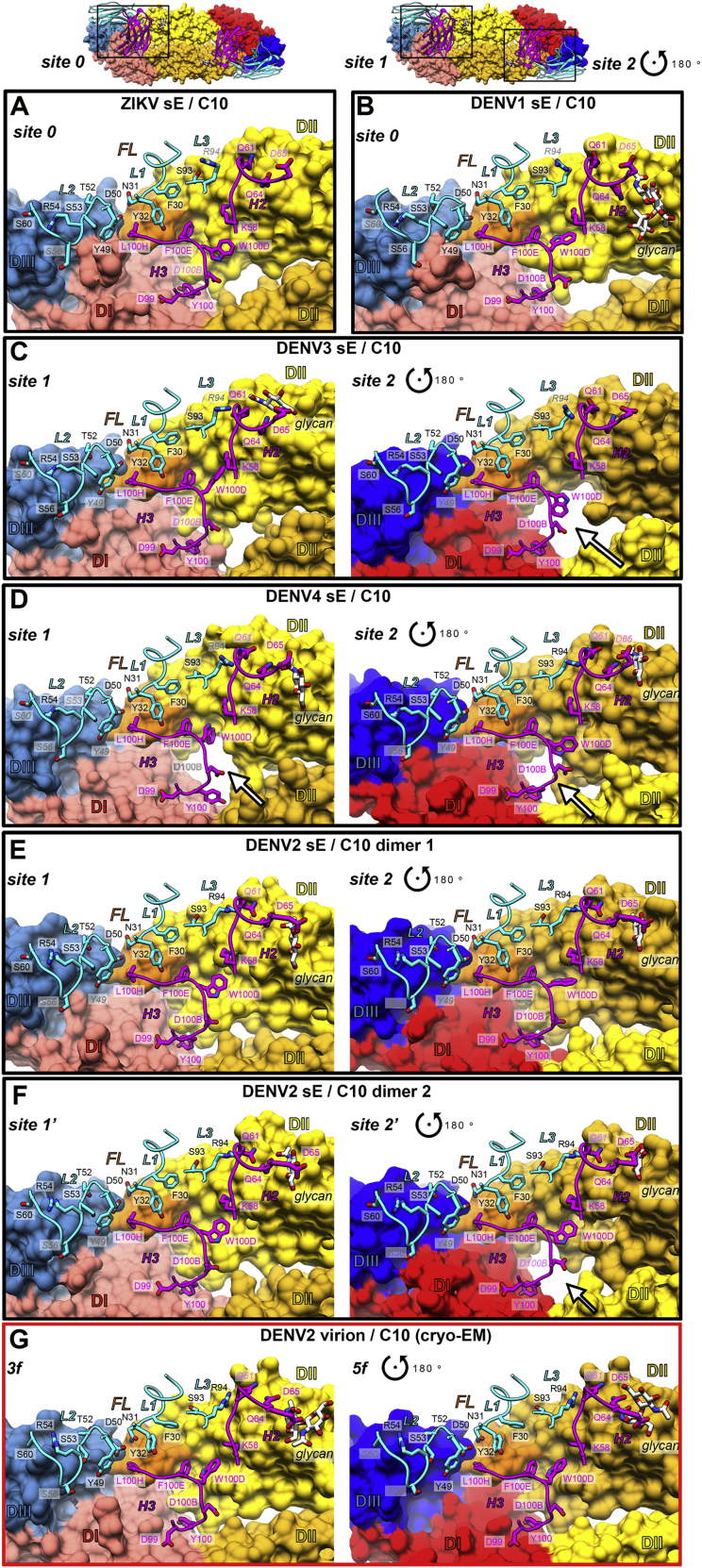
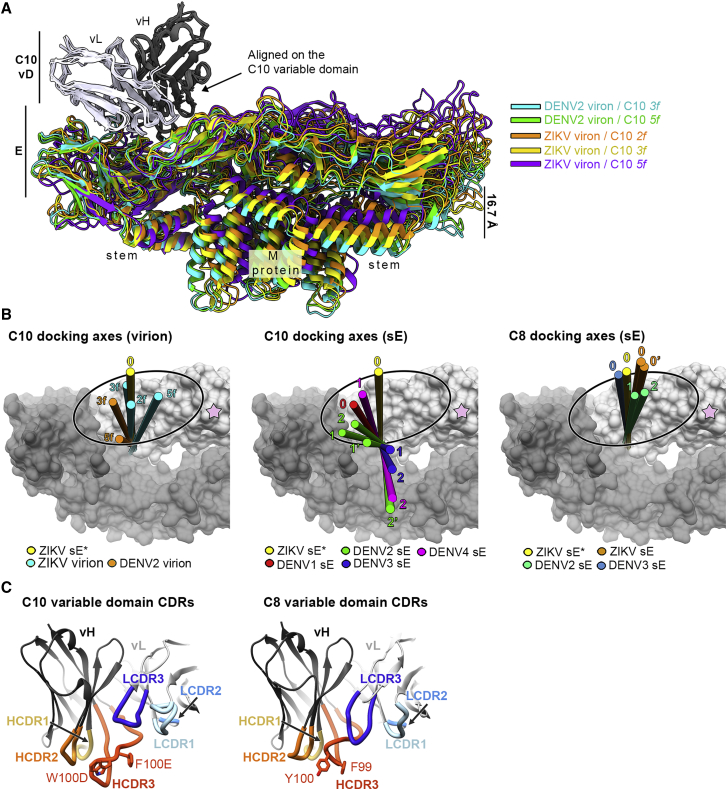
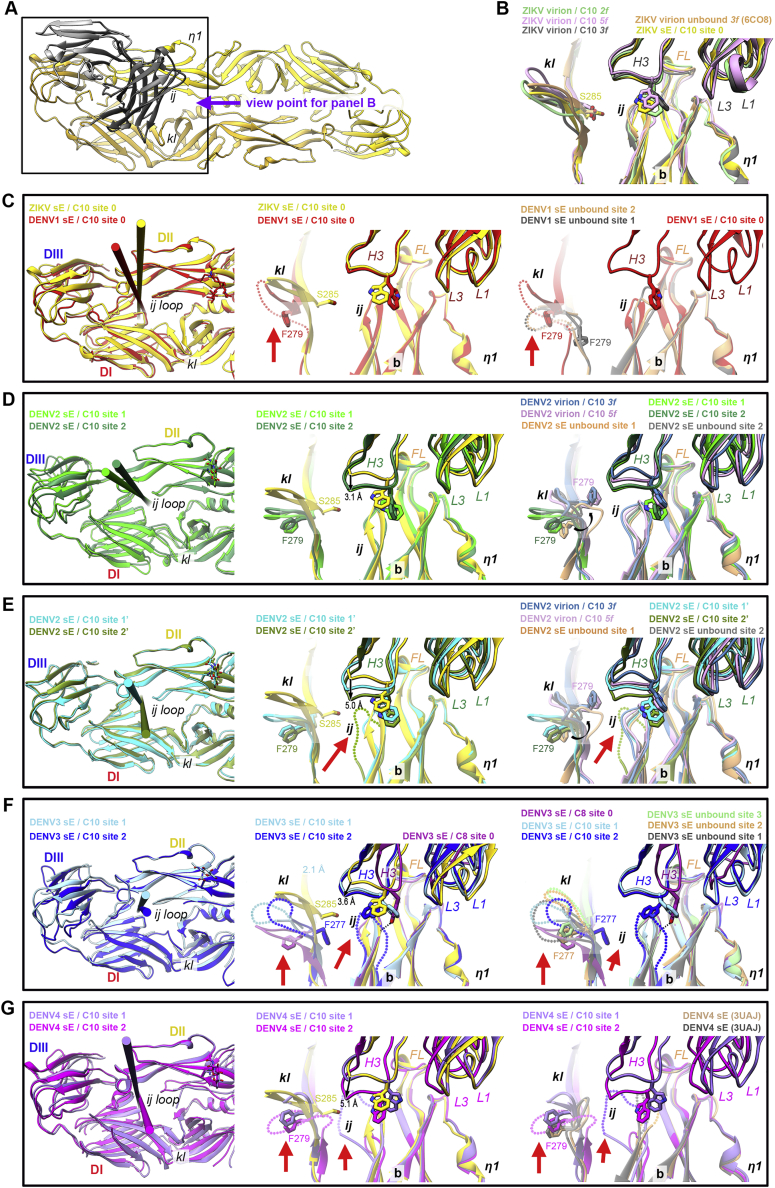
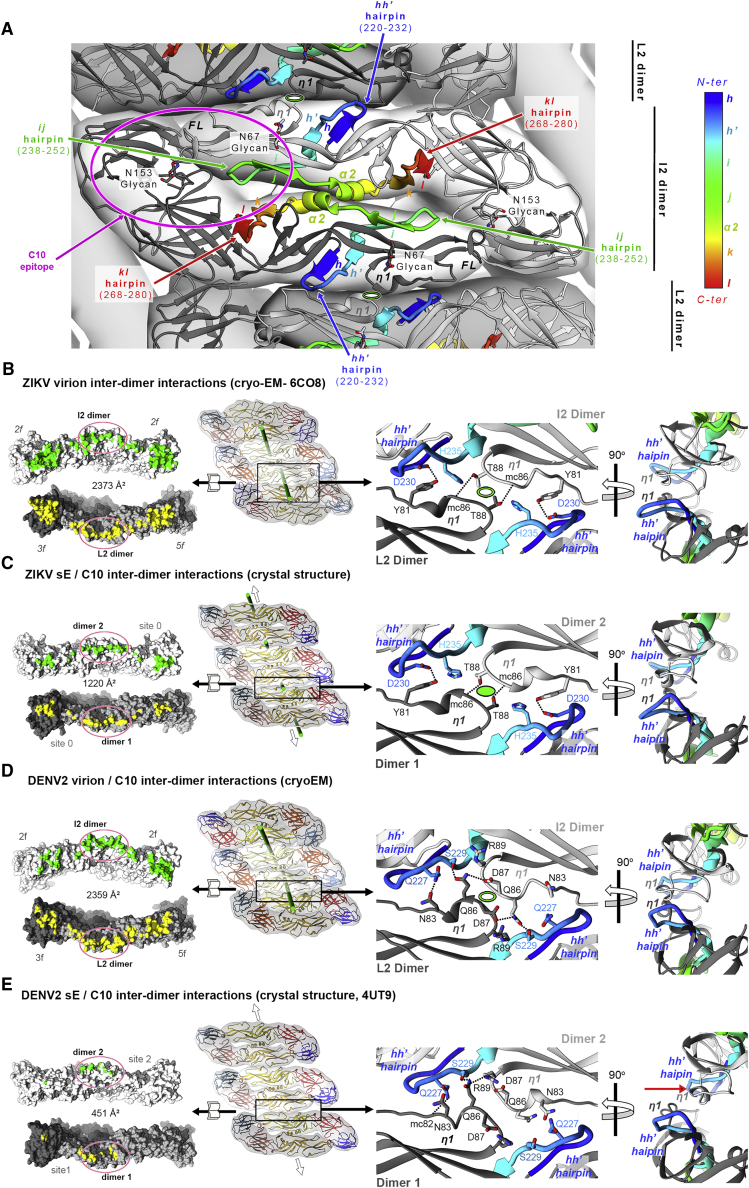
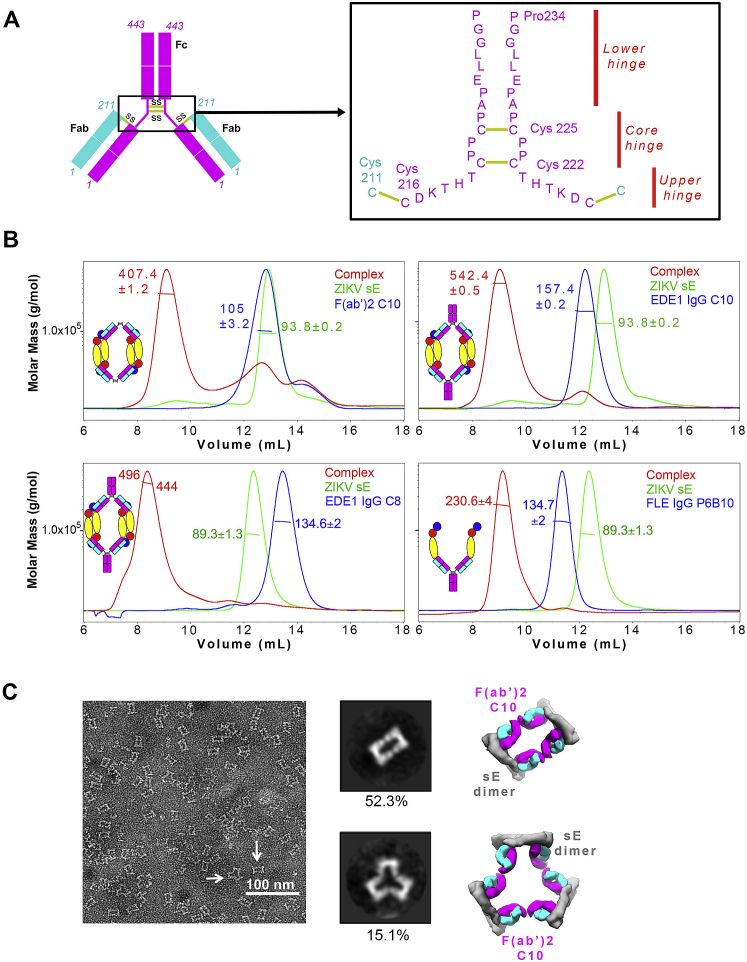
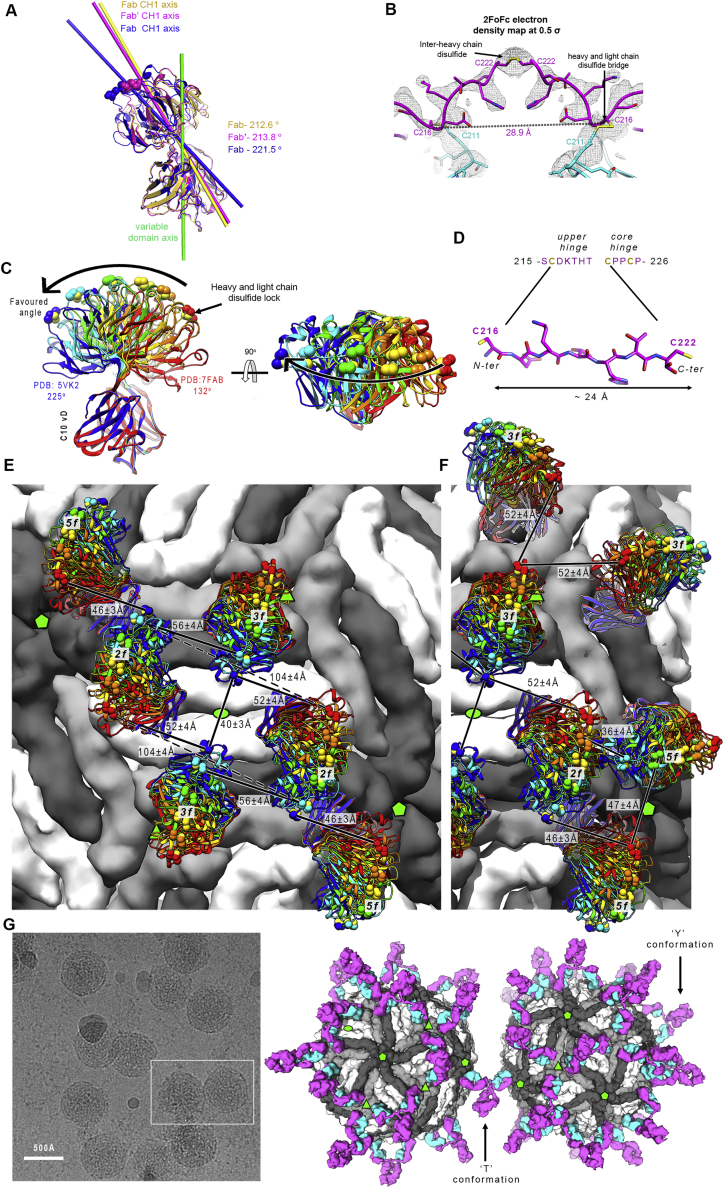
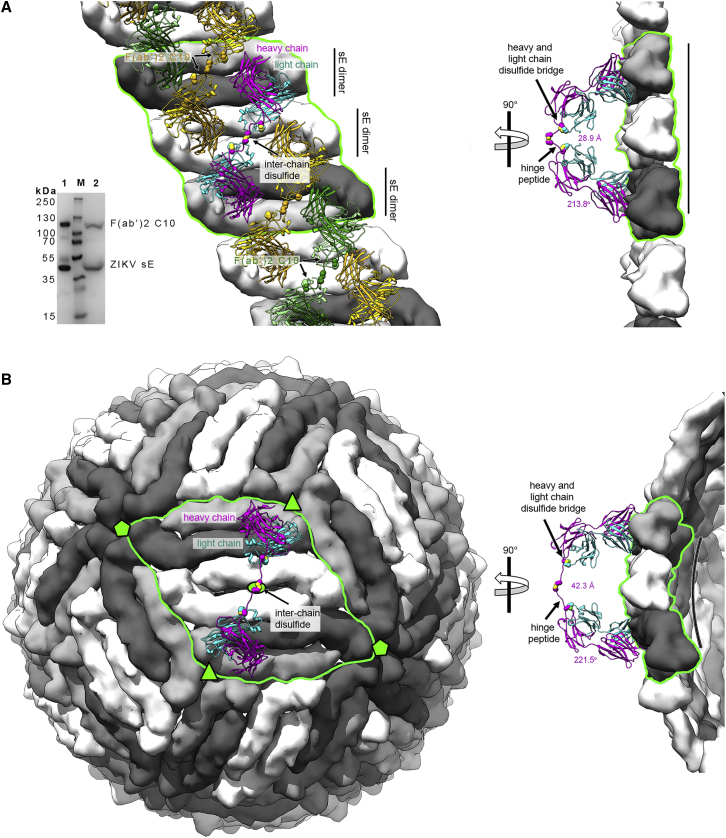
Comment in
-
How a broadly neutralizing antibody grapples with antigenic and conformational diversity in dengue virus.Cell. 2021 Dec 9;184(25):6015-6016. doi: 10.1016/j.cell.2021.11.020. Epub 2021 Dec 1. Cell. 2021. PMID: 34856127
References
-
- Barba-Spaeth G., Dejnirattisai W., Rouvinski A., Vaney M.C., Medits I., Sharma A., Simon-Lorière E., Sakuntabhai A., Cao-Lormeau V.M., Haouz A., et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature. 2016;536:48–53. - PubMed
-
- Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S.M., Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2210–2221. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
