

Tissue distribution and transcriptional regulation of CCN5 in the heart after myocardial infarction
- PMID: 34854055
- PMCID: PMC9411331
- DOI: 10.1007/s12079-021-00659-7
Tissue distribution and transcriptional regulation of CCN5 in the heart after myocardial infarction
Abstract
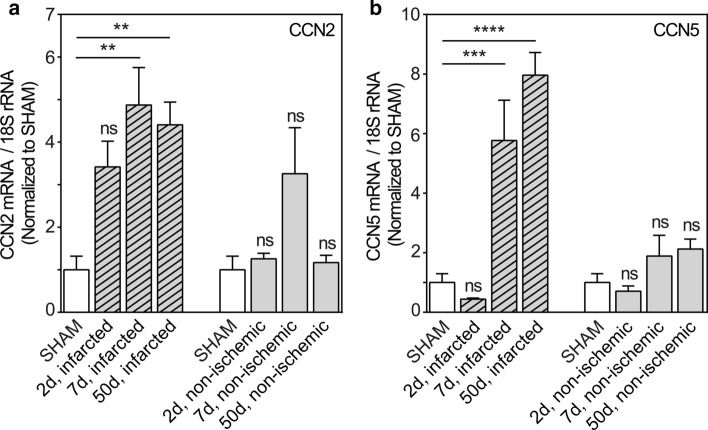
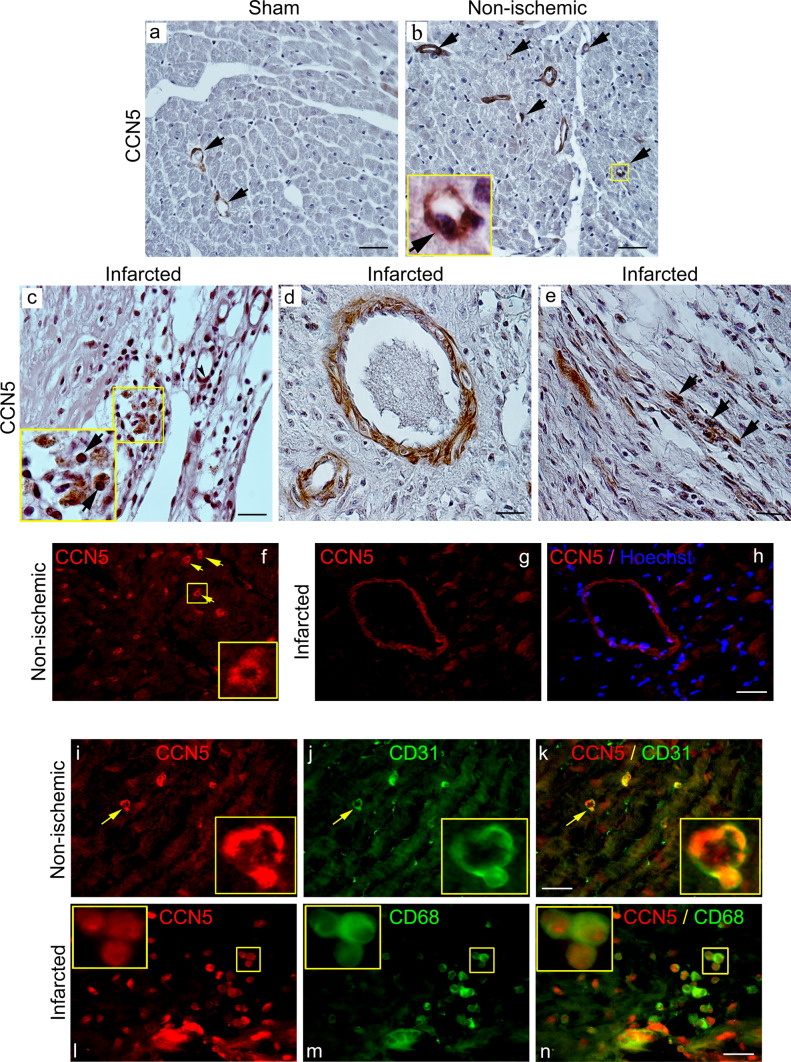
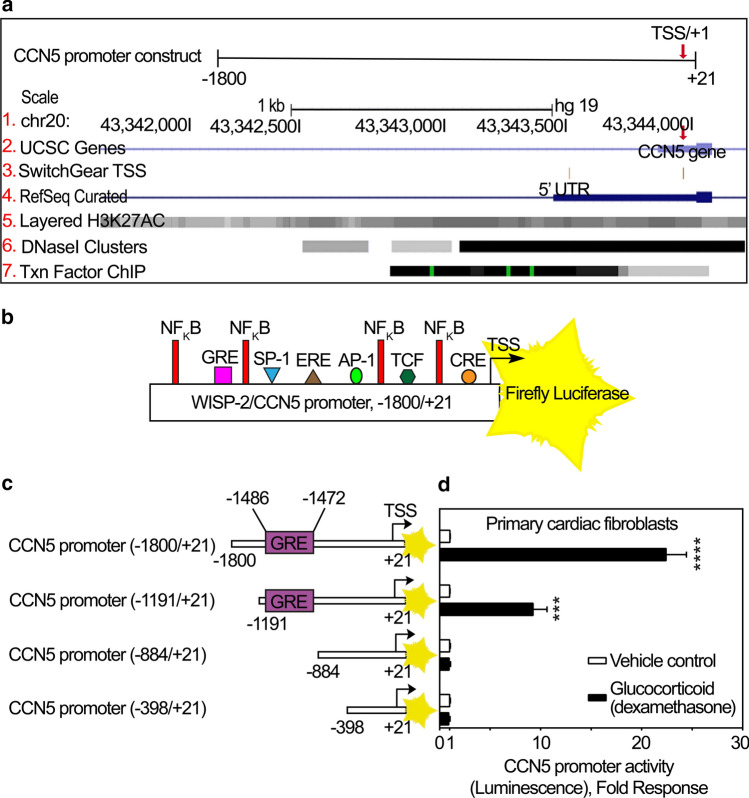
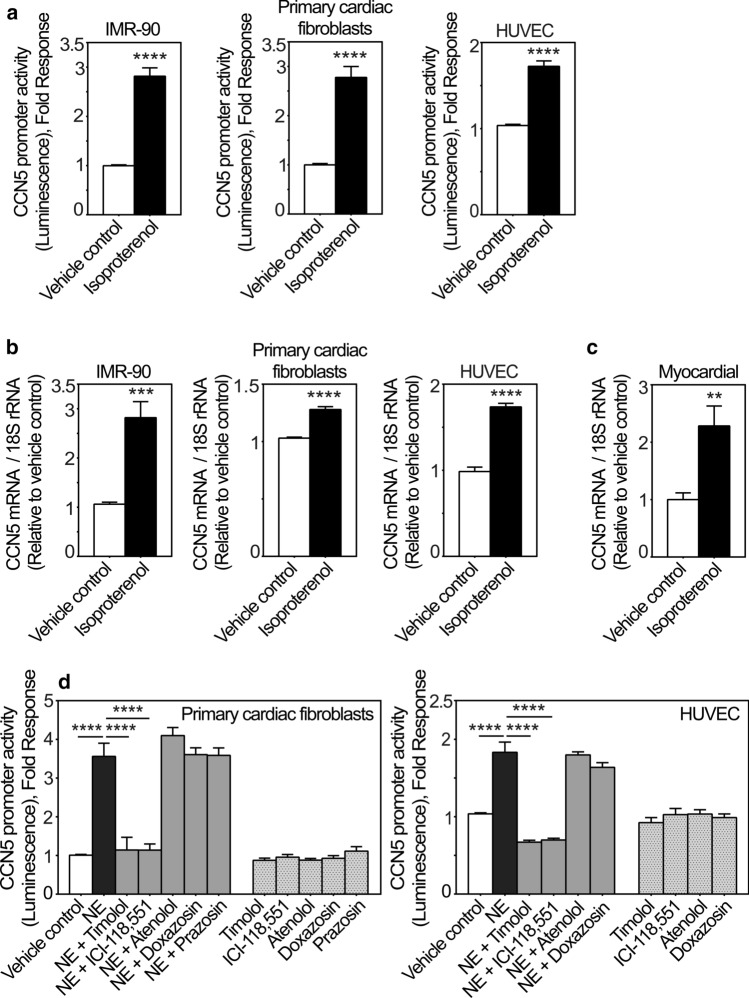
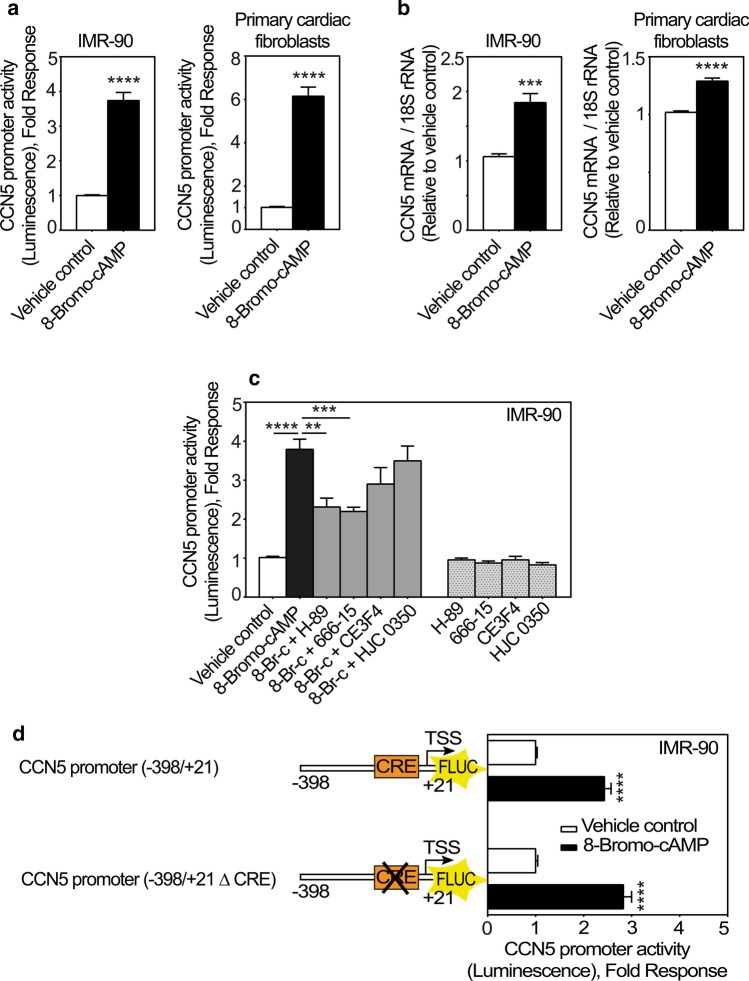
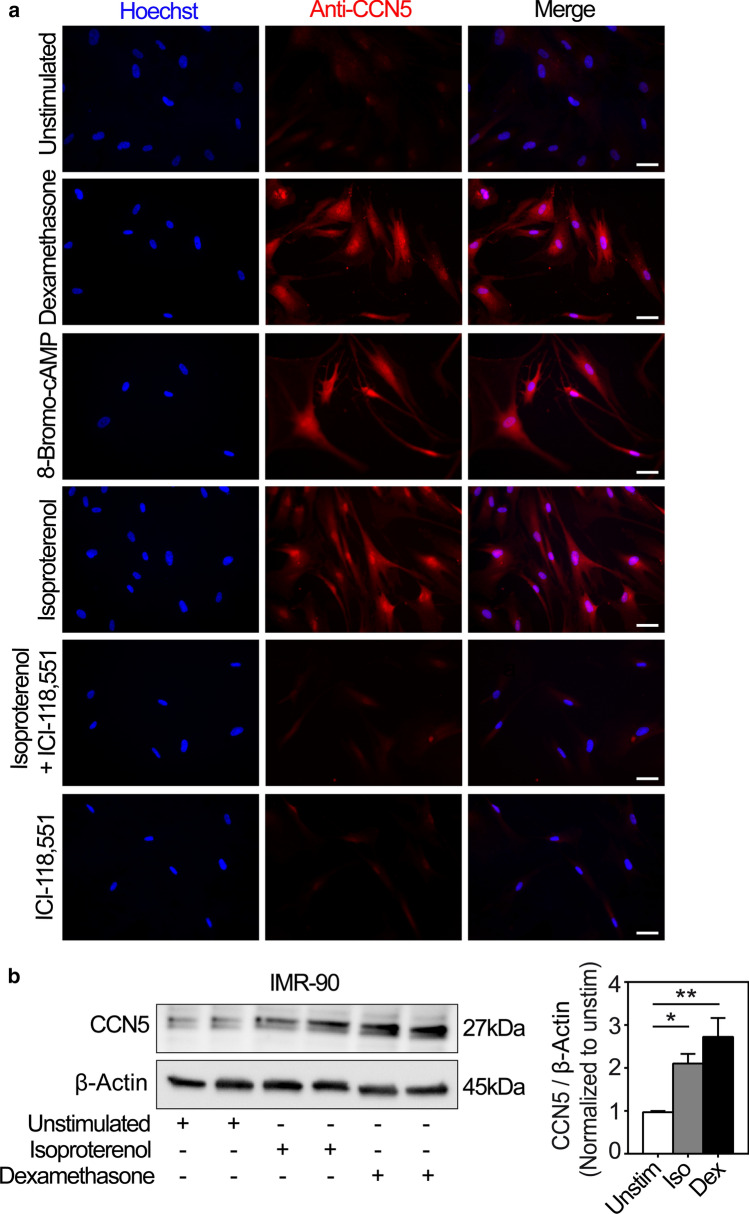
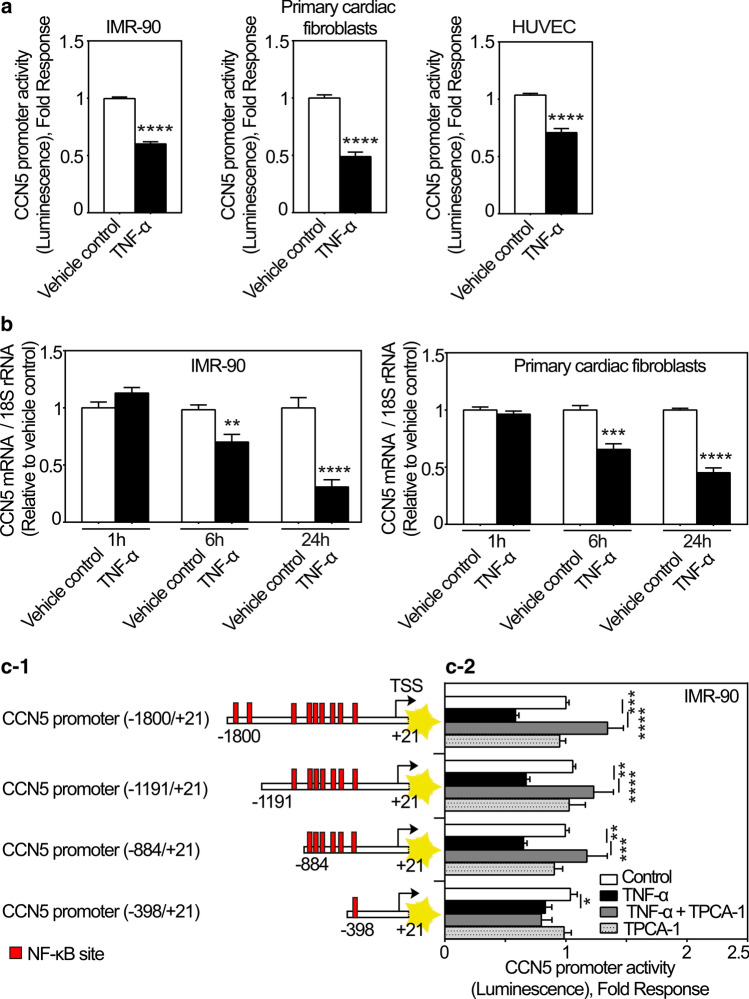
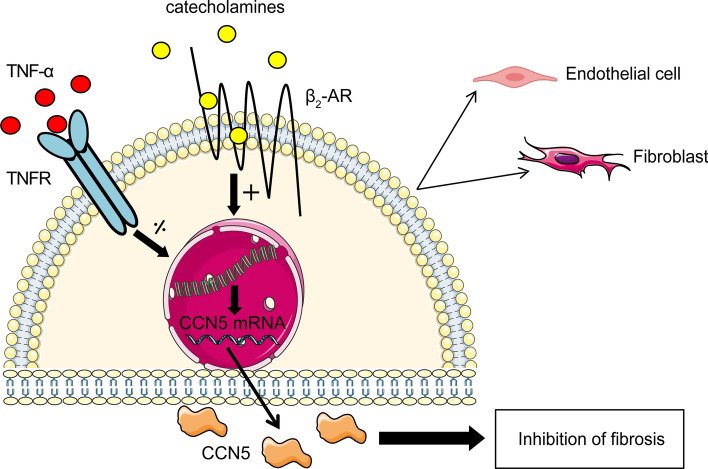
CCN5 is a divergent member of the cellular communication network factor (CCN) family in that it lacks the carboxyl terminal cystine knot domain common to the other CCN family members. CCN5 has been reported to antagonize the profibrotic actions of CCN2 and to inhibit myocardial collagen deposition and fibrosis in chronic pressure overload of the heart. However, what mechanisms that regulate CCN5 activity in the heart remain unknown. Recombinant, replication defective adenovirus encoding firefly luciferase under control of the human CCN5 promoter was prepared and used to investigate what mechanisms regulate CCN5 transcription in relevant cells. Tissue distribution of CCN5 in hearts from healthy mice and from mice subjected to myocardial infarction was investigated. Contrary to the profibrotic immediate early gene CCN2, we find that CCN5 is induced in the late proliferation and maturation phases of scar healing. CCN5 was identified principally in endothelial cells, fibroblasts, smooth muscle cells, and macrophages. Our data show that CCN5 gene transcription and protein levels are induced by catecholamines via β2-adrenergic receptors. Myocardial induction of CCN5 was further confirmed in isoproterenol-infused mice. We also find that CCN5 transcription is repressed by TNF-α, an inflammatory mediator highly elevated in early phases of wound healing following myocardial infarction. In conclusion, CCN5 predominates in endothelial cells, fibroblasts, and macrophages of the differentiating scar tissue and its transcription is conversely regulated by β2-adrenergic agonists and TNF-α.
Keywords: CCN2; CCN5; Catecholamines; Myocardial infarction; Primary cardiac fibroblasts; β2-Adrenergic receptor.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
The carboxyl-terminal TSP1-homology domain is the biologically active effector peptide of matricellular protein CCN5 that counteracts profibrotic CCN2.J Biol Chem. 2023 Jan;299(1):102803. doi: 10.1016/j.jbc.2022.102803. Epub 2022 Dec 15. J Biol Chem. 2023. PMID: 36529291 Free PMC article.
-
CCN5 overexpression inhibits profibrotic phenotypes via the PI3K/Akt signaling pathway in lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis and in an in vivo model of lung fibrosis.Int J Mol Med. 2014 Feb;33(2):478-86. doi: 10.3892/ijmm.2013.1565. Epub 2013 Nov 25. Int J Mol Med. 2014. PMID: 24276150
-
CCN2 and CCN5 exerts opposing effect on fibroblast proliferation and transdifferentiation induced by TGF-β.Clin Exp Pharmacol Physiol. 2015 Nov;42(11):1207-19. doi: 10.1111/1440-1681.12470. Clin Exp Pharmacol Physiol. 2015. PMID: 26218313
-
Role of CCN5 (WNT1 inducible signaling pathway protein 2) in pancreatic islets.J Diabetes. 2017 May;9(5):462-474. doi: 10.1111/1753-0407.12507. Epub 2016 Dec 26. J Diabetes. 2017. PMID: 27863006 Review.
-
Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice.Int J Mol Sci. 2021 Dec 14;22(24):13418. doi: 10.3390/ijms222413418. Int J Mol Sci. 2021. PMID: 34948212 Free PMC article. Review.
Cited by
-
CCN5 negatively regulates TGF-β-induced endometriosis associated fibrosis through Wnt/β-catenin signaling via Smad3-dependent mechanism.J Transl Med. 2025 Jul 10;23(1):763. doi: 10.1186/s12967-025-06804-9. J Transl Med. 2025. PMID: 40640862 Free PMC article.
-
CCN5/WISP2 serum levels in patients with coronary artery disease and type 2 diabetes and its correlation with inflammation and insulin resistance; a machine learning approach.Biochem Biophys Rep. 2024 Oct 30;40:101857. doi: 10.1016/j.bbrep.2024.101857. eCollection 2024 Dec. Biochem Biophys Rep. 2024. PMID: 39552711 Free PMC article.
-
Modeling the microenvironment special issue.J Cell Commun Signal. 2022 Dec;16(4):631-632. doi: 10.1007/s12079-022-00675-1. Epub 2022 Mar 29. J Cell Commun Signal. 2022. PMID: 35352286 Free PMC article. No abstract available.
-
CCN proteins: opportunities for clinical studies-a personal perspective.J Cell Commun Signal. 2023 Jun;17(2):333-352. doi: 10.1007/s12079-023-00761-y. Epub 2023 May 17. J Cell Commun Signal. 2023. PMID: 37195381 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
