

Integrative epigenomics in Sjögren´s syndrome reveals novel pathways and a strong interaction between the HLA, autoantibodies and the interferon signature
- PMID: 34857786
- PMCID: PMC8640069
- DOI: 10.1038/s41598-021-01324-0
Integrative epigenomics in Sjögren´s syndrome reveals novel pathways and a strong interaction between the HLA, autoantibodies and the interferon signature
Abstract
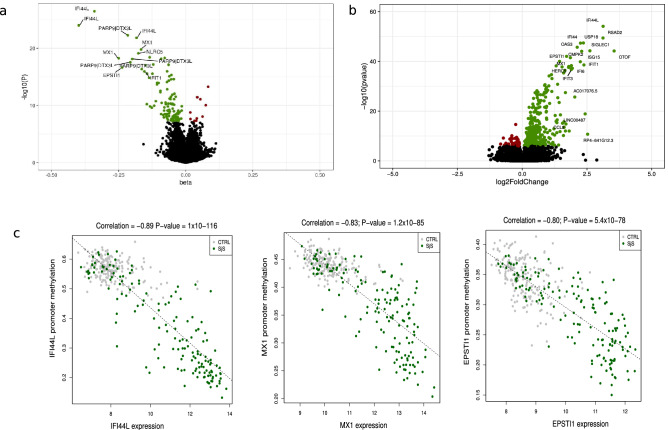
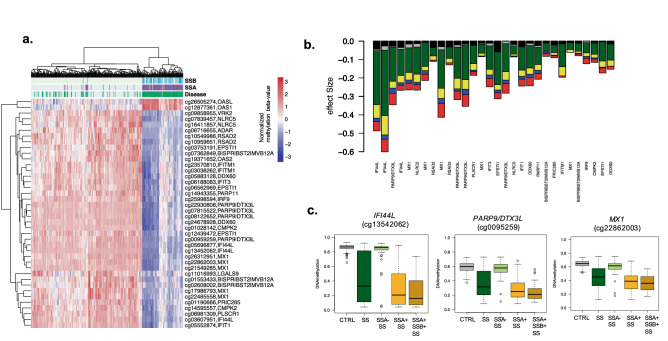
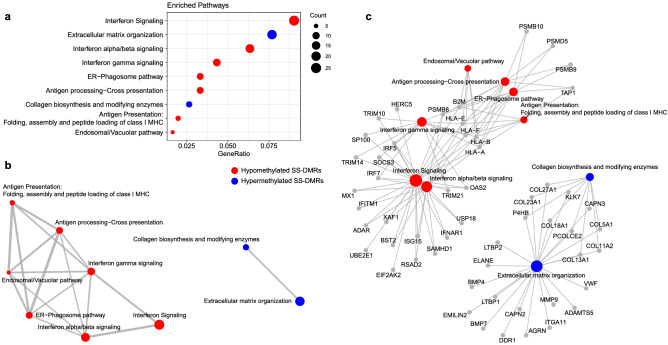
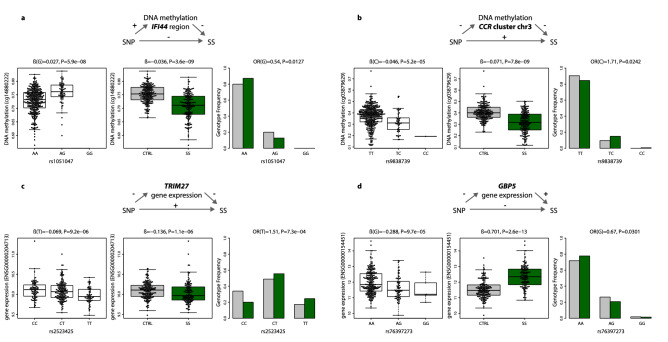
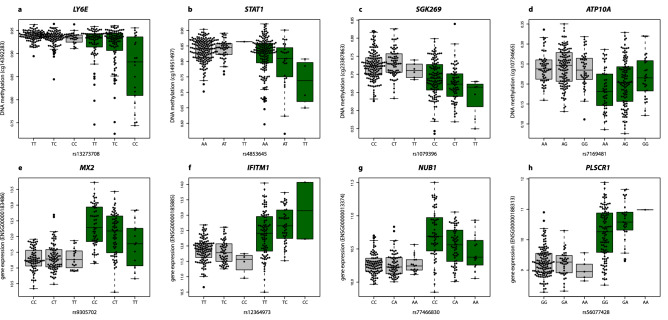
Primary Sjögren's syndrome (SS) is a systemic autoimmune disease characterized by lymphocytic infiltration and damage of exocrine salivary and lacrimal glands. The etiology of SS is complex with environmental triggers and genetic factors involved. By conducting an integrated multi-omics study, we confirmed a vast coordinated hypomethylation and overexpression effects in IFN-related genes, what is known as the IFN signature. Stratified and conditional analyses suggest a strong interaction between SS-associated HLA genetic variation and the presence of Anti-Ro/SSA autoantibodies in driving the IFN epigenetic signature and determining SS. We report a novel epigenetic signature characterized by increased DNA methylation levels in a large number of genes enriched in pathways such as collagen metabolism and extracellular matrix organization. We identified potential new genetic variants associated with SS that might mediate their risk by altering DNA methylation or gene expression patterns, as well as disease-interacting genetic variants that exhibit regulatory function only in the SS population. Our study sheds new light on the interaction between genetics, autoantibody profiles, DNA methylation and gene expression in SS, and contributes to elucidate the genetic architecture of gene regulation in an autoimmune population.
© 2021. The Author(s).
Conflict of interest statement
Zuzanna Makowska and Anne Buttgereit are employees of BAYER AG. All other authors have no competing interests.
Figures
References
-
- Fox RI. Sjögren’s syndrome. Lancet. 2005;366:321–331. - PubMed
-
- Rischmueller M, Tieu J, Lester S. Primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2016;30:189–220. - PubMed
-
- Teruel M, Alarcón-Riquelme ME. Genetics of systemic lupus erythematosus and Sjögren’s syndrome: An update. Curr. Opin. Rheumatol. 2016;28:506–514. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
