

In vitro interaction network of a synthetic gut bacterial community
- PMID: 34857933
- PMCID: PMC8941000
- DOI: 10.1038/s41396-021-01153-z
In vitro interaction network of a synthetic gut bacterial community
Abstract
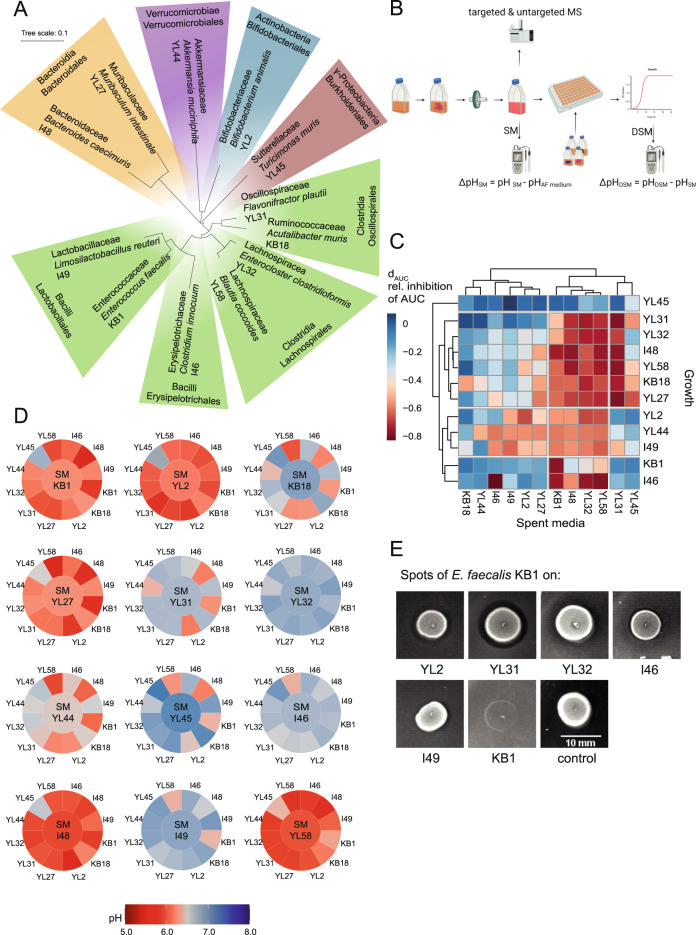
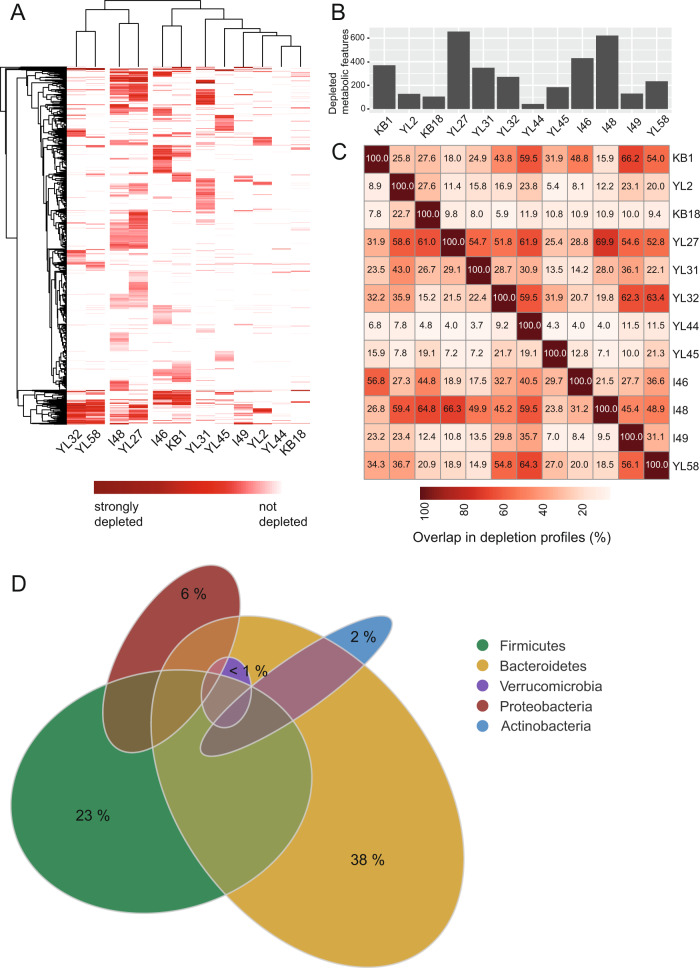
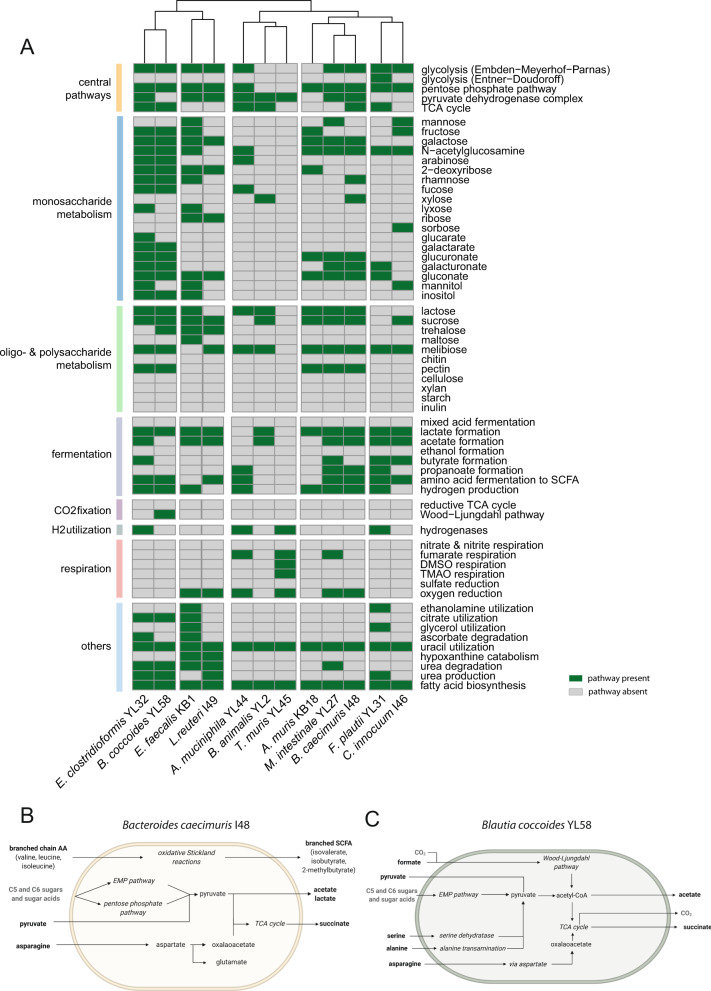
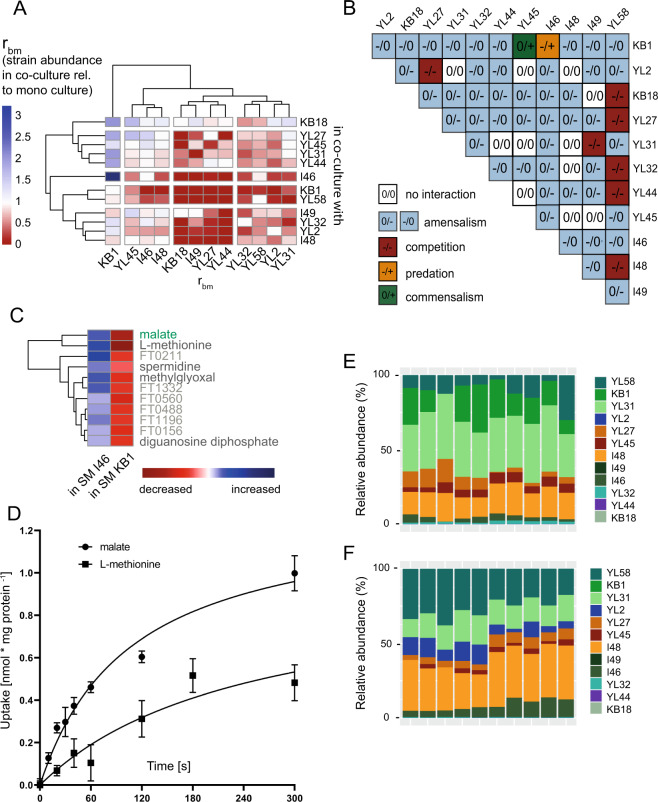
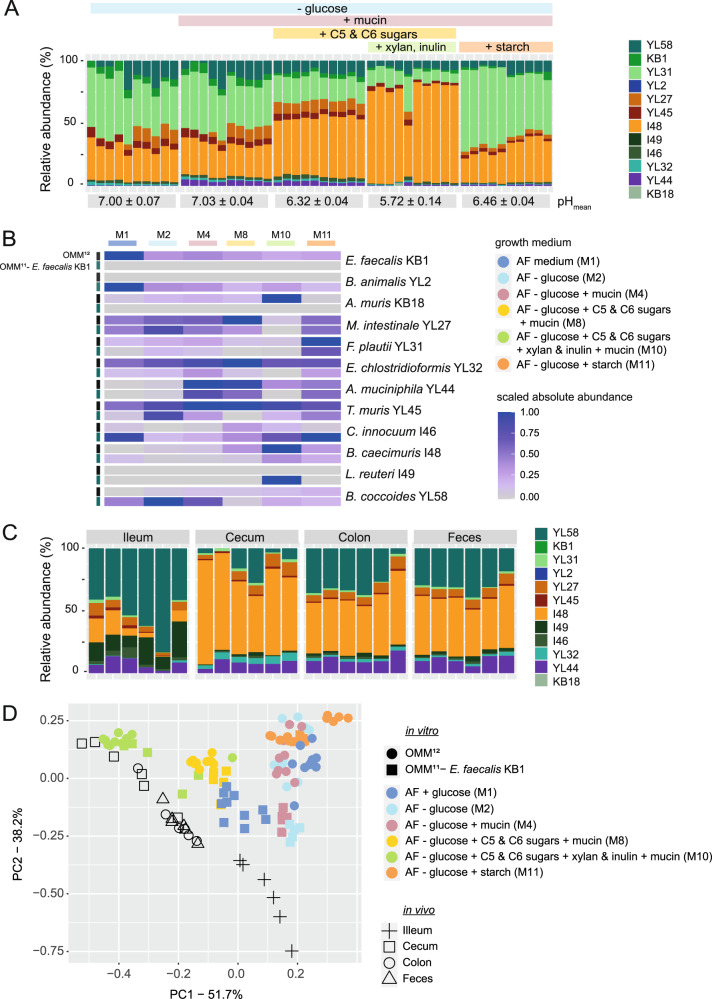
A key challenge in microbiome research is to predict the functionality of microbial communities based on community membership and (meta)-genomic data. As central microbiota functions are determined by bacterial community networks, it is important to gain insight into the principles that govern bacteria-bacteria interactions. Here, we focused on the growth and metabolic interactions of the Oligo-Mouse-Microbiota (OMM12) synthetic bacterial community, which is increasingly used as a model system in gut microbiome research. Using a bottom-up approach, we uncovered the directionality of strain-strain interactions in mono- and pairwise co-culture experiments as well as in community batch culture. Metabolic network reconstruction in combination with metabolomics analysis of bacterial culture supernatants provided insights into the metabolic potential and activity of the individual community members. Thereby, we could show that the OMM12 interaction network is shaped by both exploitative and interference competition in vitro in nutrient-rich culture media and demonstrate how community structure can be shifted by changing the nutritional environment. In particular, Enterococcus faecalis KB1 was identified as an important driver of community composition by affecting the abundance of several other consortium members in vitro. As a result, this study gives fundamental insight into key drivers and mechanistic basis of the OMM12 interaction network in vitro, which serves as a knowledge base for future mechanistic in vivo studies.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 395357507/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 315980449/Deutsche Forschungsgemeinschaft (German Research Foundation)
- CRC 1182/Deutsche Forschungsgemeinschaft (German Research Foundation)
- Cluster of Excellence 2167/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 865615/EC | EC Seventh Framework Programm | FP7 Ideas: European Research Council (FP7-IDEAS-ERC - Specific Programme: "Ideas" Implementing the Seventh Framework Programme of the European Community for Research, Technological Development and Demonstration Activities (2007 to 2013))
LinkOut - more resources
Full Text Sources
Research Materials
