

PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments
- PMID: 34859208
- PMCID: PMC8632735
- DOI: 10.1093/nargab/lqab101
PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments
Abstract
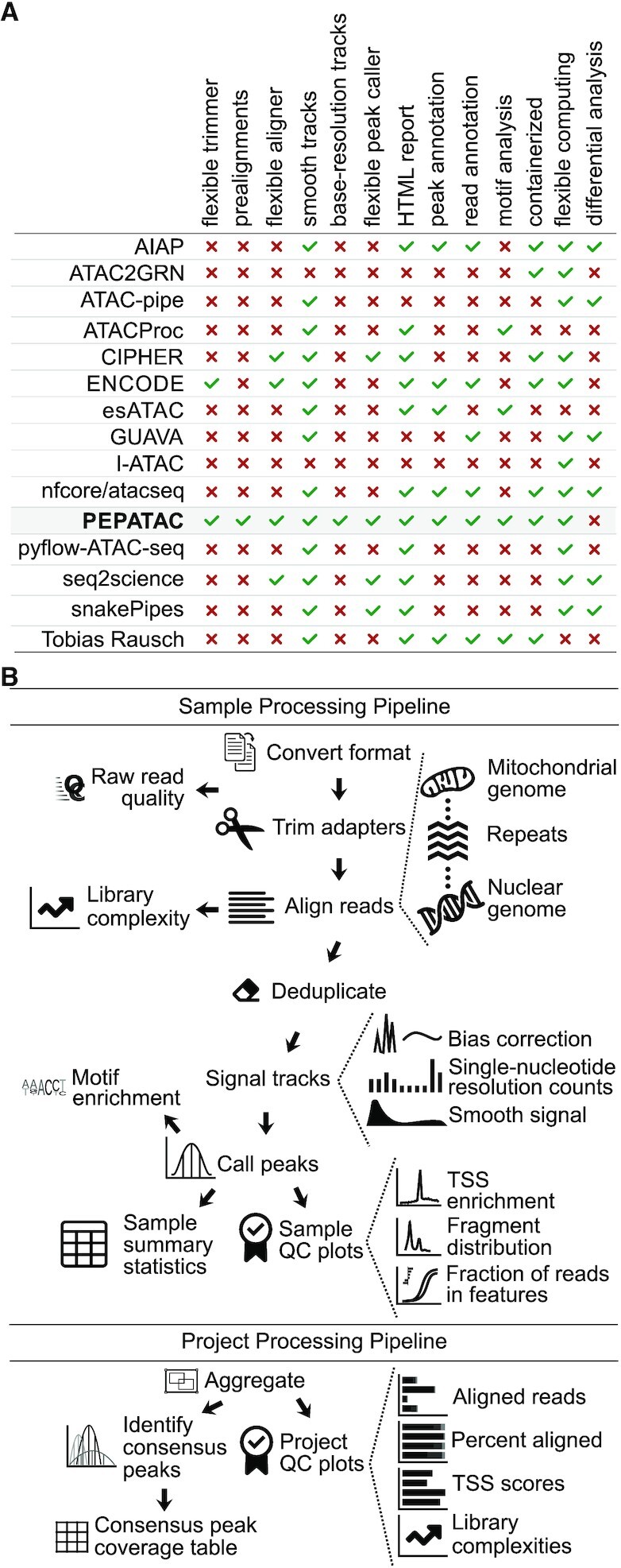
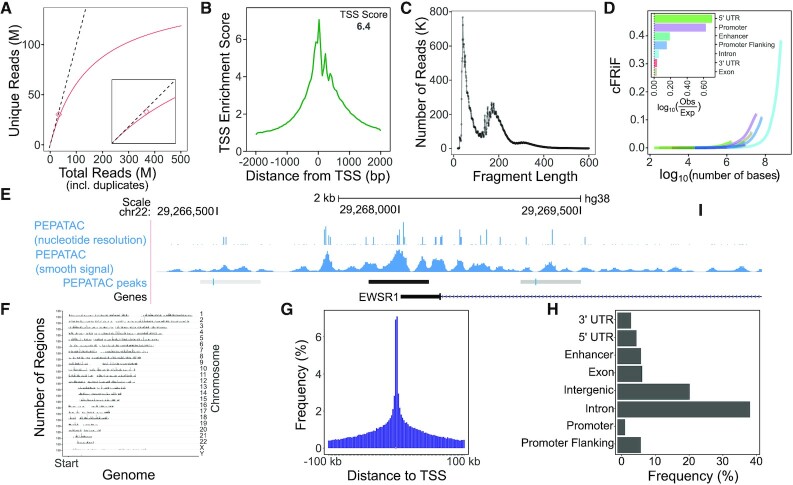
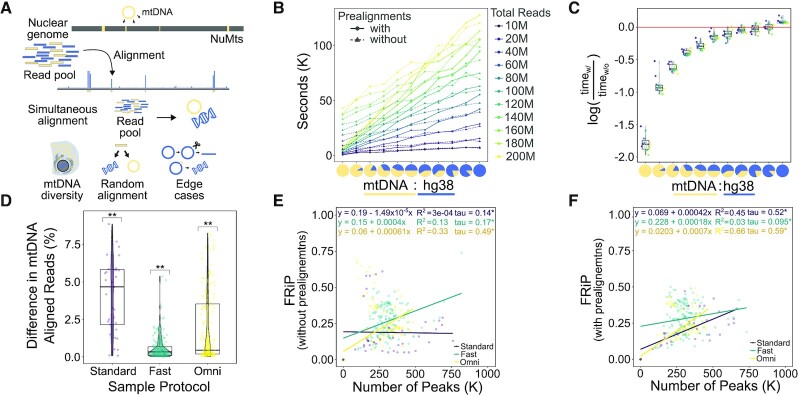
As chromatin accessibility data from ATAC-seq experiments continues to expand, there is continuing need for standardized analysis pipelines. Here, we present PEPATAC, an ATAC-seq pipeline that is easily applied to ATAC-seq projects of any size, from one-off experiments to large-scale sequencing projects. PEPATAC leverages unique features of ATAC-seq data to optimize for speed and accuracy, and it provides several unique analytical approaches. Output includes convenient quality control plots, summary statistics, and a variety of generally useful data formats to set the groundwork for subsequent project-specific data analysis. Downstream analysis is simplified by a standard definition format, modularity of components, and metadata APIs in R and Python. It is restartable, fault-tolerant, and can be run on local hardware, using any cluster resource manager, or in provided Linux containers. We also demonstrate the advantage of aligning to the mitochondrial genome serially, which improves the accuracy of alignment statistics and quality control metrics. PEPATAC is a robust and portable first step for any ATAC-seq project. BSD2-licensed code and documentation are available at https://pepatac.databio.org.
© The Author(s) 2021. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
-
- Sheffield N.C., Thurman R.E., Song L., Safi A., Stamatoyannopoulos J.A., Lenhard B., Crawford G.E., Furey T.S. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013; 23:777–788. - PMC - PubMed
