

Facioscapulohumeral dystrophy weakened sarcomeric contractility is mimicked in induced pluripotent stem cells-derived innervated muscle fibres
- PMID: 34859613
- PMCID: PMC8818656
- DOI: 10.1002/jcsm.12835
Facioscapulohumeral dystrophy weakened sarcomeric contractility is mimicked in induced pluripotent stem cells-derived innervated muscle fibres
Abstract
Background: Facioscapulohumeral dystrophy (FSHD) is a late-onset autosomal dominant form of muscular dystrophy involving specific groups of muscles with variable weakness that precedes inflammatory response, fat infiltration, and muscle atrophy. As there is currently no cure for this disease, understanding and modelling the typical muscle weakness in FSHD remains a major milestone towards deciphering the disease pathogenesis as it will pave the way to therapeutic strategies aimed at correcting the functional muscular defect in patients.
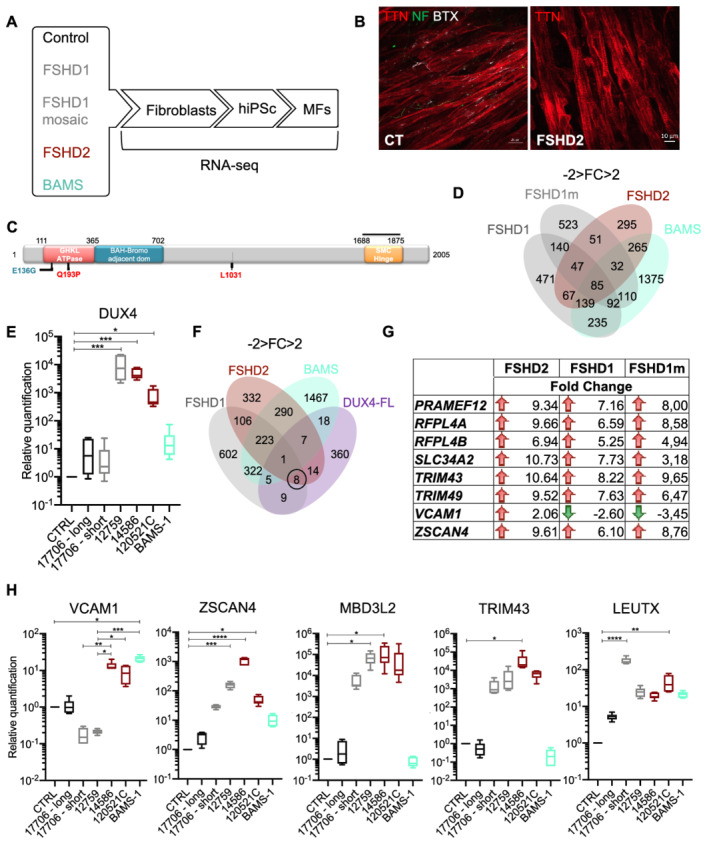
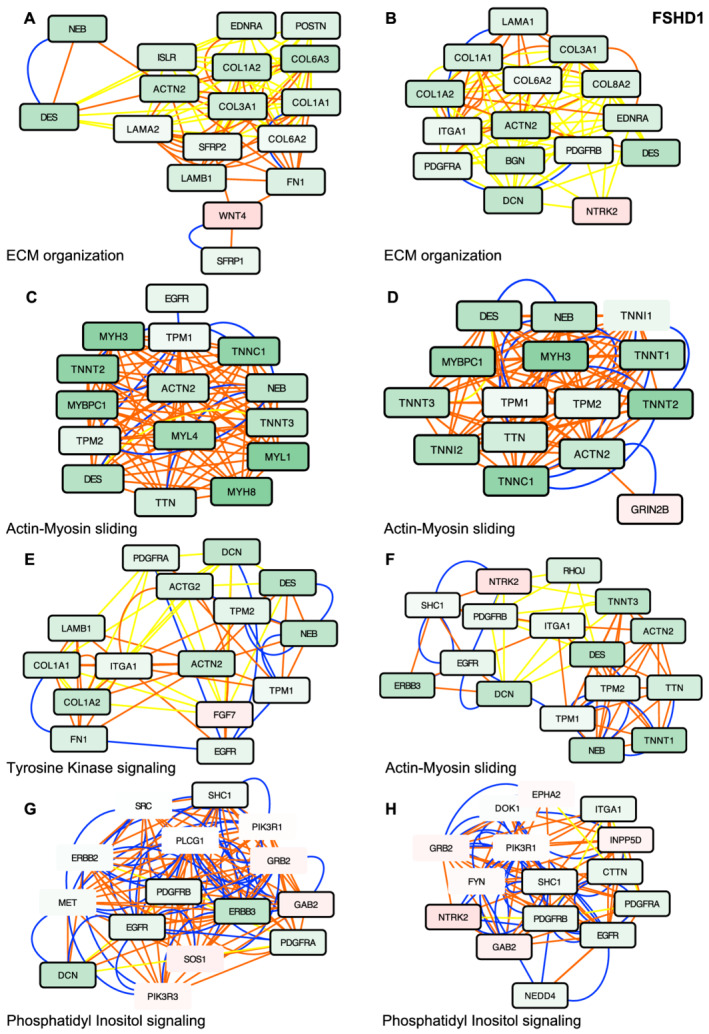
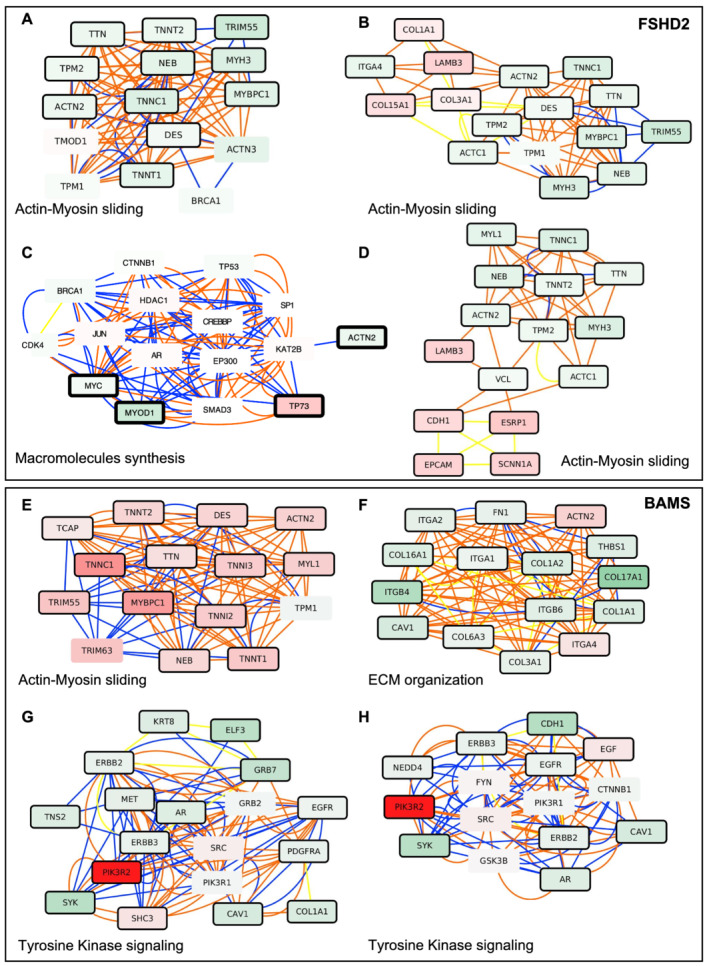
Methods: To gain further insights into the specificity of the muscle alteration in this disease, we derived induced pluripotent stem cells from patients affected with Types 1 and 2 FSHD but also from patients affected with Bosma arhinia and microphthalmia. We differentiated these cells into contractile innervated muscle fibres and analysed their transcriptome by RNA Seq in comparison with cells derived from healthy donors. To uncover biological pathways altered in the disease, we applied MOGAMUN, a multi-objective genetic algorithm that integrates multiplex complex networks of biological interactions (protein-protein interactions, co-expression, and biological pathways) and RNA Seq expression data to identify active modules.
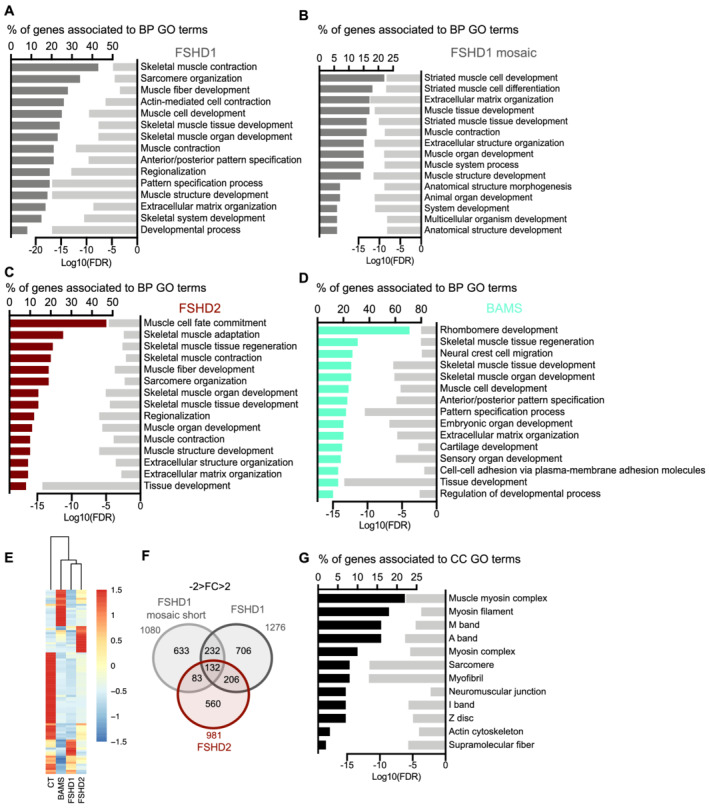
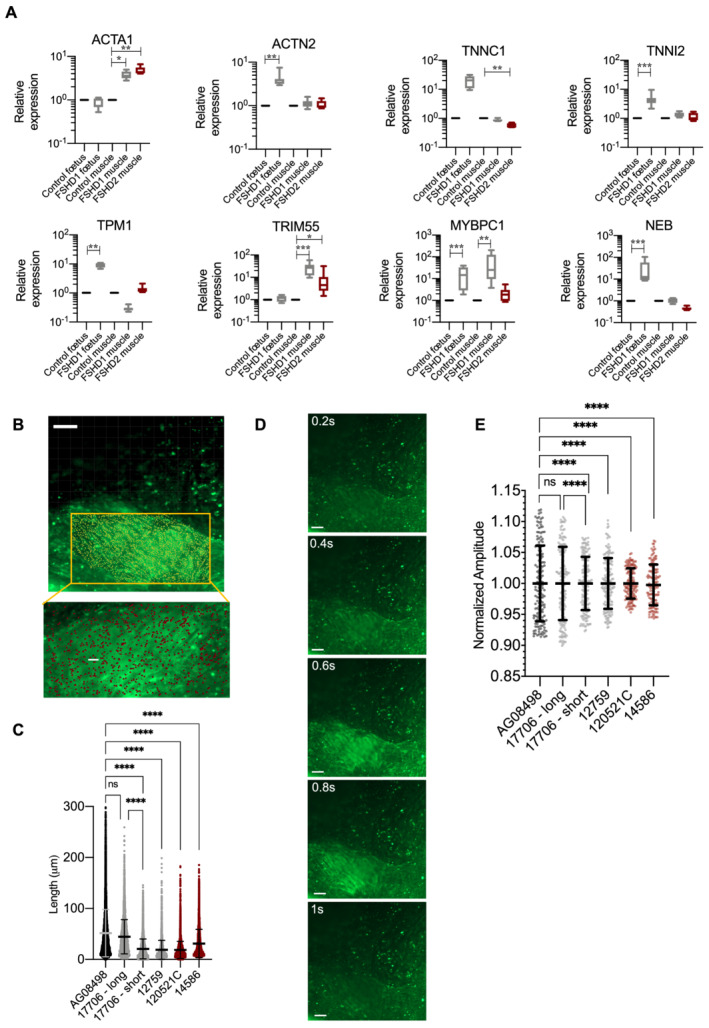
Results: We identified 132 differentially expressed genes that are specific to FSHD cells (false discovery rate < 0.05). In FSHD, the vast majority of active modules retrieved with MOGAMUN converges towards a decreased expression of genes encoding proteins involved in sarcomere organization (P value 2.63e-12 ), actin cytoskeleton (P value 9.4e-5 ), myofibril (P value 2.19e-12 ), actin-myosin sliding, and calcium handling (with P values ranging from 7.9e-35 to 7.9e-21 ). Combined with in vivo validations and functional investigations, our data emphasize a reduction in fibre contraction (P value < 0.0001) indicating that the muscle weakness that is typical of FSHD clinical spectrum might be associated with dysfunction of calcium release (P value < 0.0001), actin-myosin interactions, motor activity, mechano-transduction, and dysfunctional sarcomere contractility.
Conclusions: Identification of biomarkers of FSHD muscle remain critical for understanding the process leading to the pathology but also for the definition of readouts to be used for drug design, outcome measures, and monitoring of therapies. The different pathways identified through a system biology approach have been largely overlooked in the disease. Overall, our work opens new perspectives in the definition of biomarkers able to define the muscle alteration but also in the development of novel strategies to improve muscle function as it provides functional parameters for active molecule screening.
Keywords: Facioscapulohumeral dystrophy; Induced pluripotent stem cells; Muscle contraction; Muscle weakening; Pathophysiology; Sarcomere; System biology.
© 2021 The Authors. Journal of Cachexia, Sarcopenia and Muscle published by John Wiley & Sons Ltd on behalf of Society on Sarcopenia, Cachexia and Wasting Disorders.
Conflict of interest statement
No conflict of interest declared.
Figures
References
-
- Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy. Curr Opin Neurol 2009;22:539–542. - PubMed
-
- Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north‐east Italian population sample. Clin Genet 2009;75:550–555. - PubMed
-
- Lemmers RJ, de Kievit P, Sandkuijl L, Padberg GW, van Ommen GJ, Frants RR, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet 2002;32:235–236. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
