

Clean room microbiome complexity impacts planetary protection bioburden
- PMID: 34861887
- PMCID: PMC8643001
- DOI: 10.1186/s40168-021-01159-x
Clean room microbiome complexity impacts planetary protection bioburden
Abstract
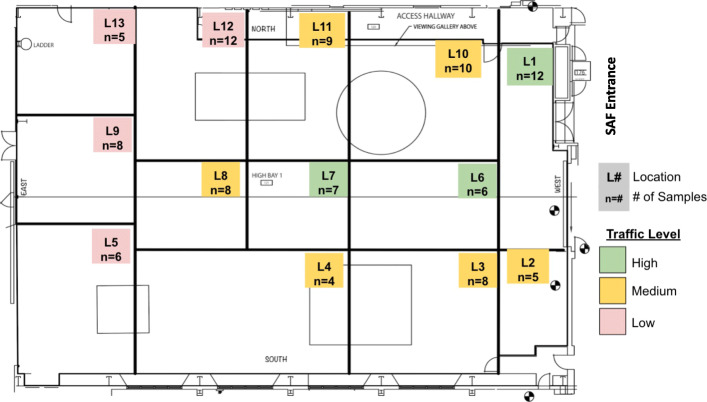
Background: The Spacecraft Assembly Facility (SAF) at the NASA's Jet Propulsion Laboratory is the primary cleanroom facility used in the construction of some of the planetary protection (PP)-sensitive missions developed by NASA, including the Mars 2020 Perseverance Rover that launched in July 2020. SAF floor samples (n=98) were collected, over a 6-month period in 2016 prior to the construction of the Mars rover subsystems, to better understand the temporal and spatial distribution of bacterial populations (total, viable, cultivable, and spore) in this unique cleanroom.
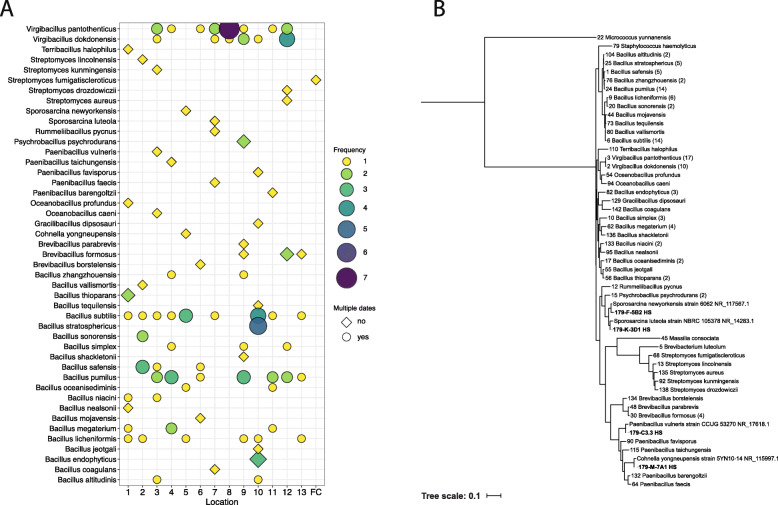
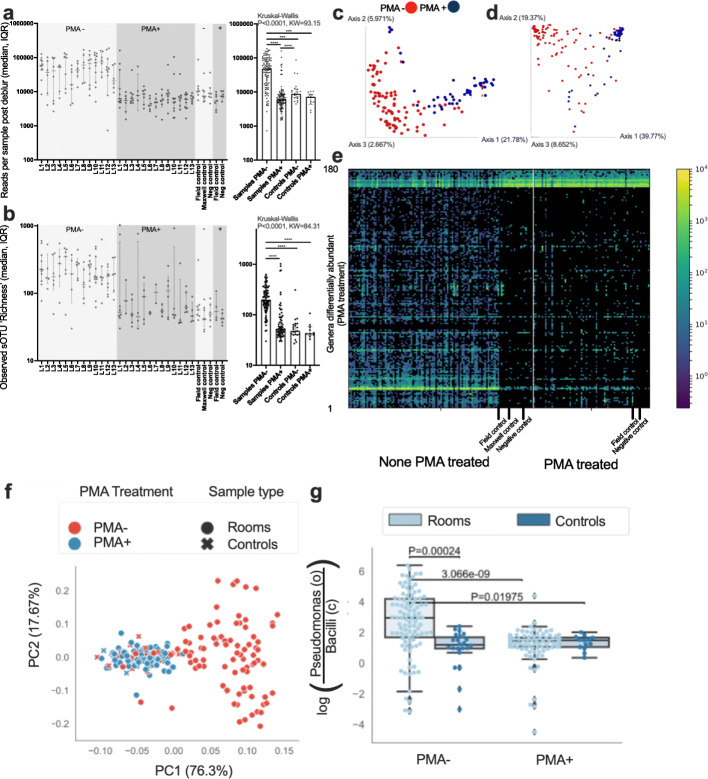
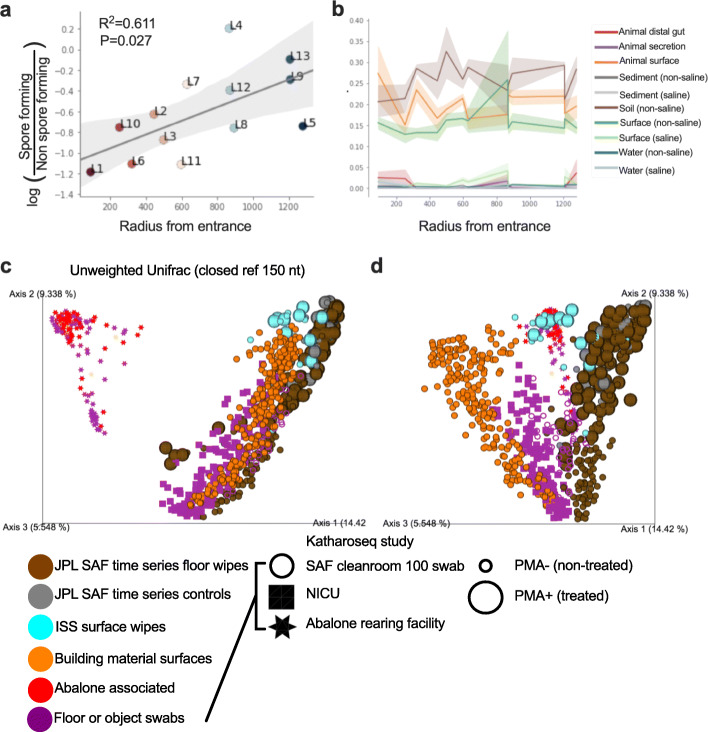
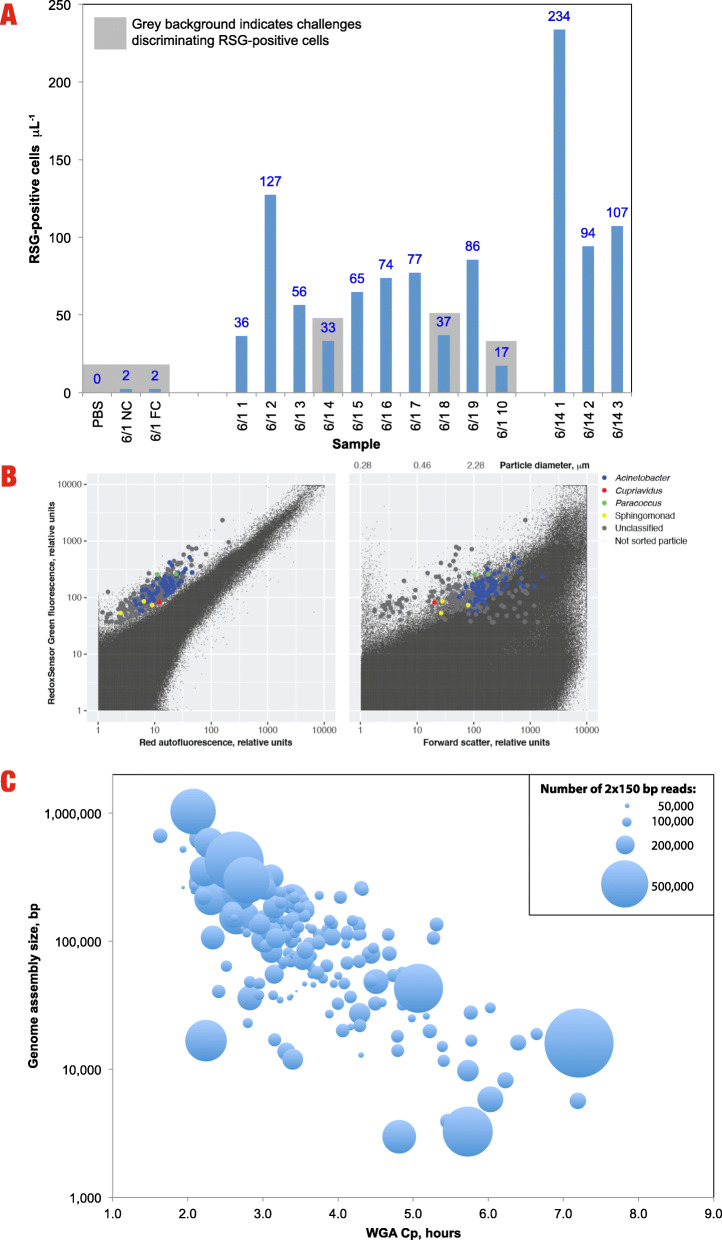
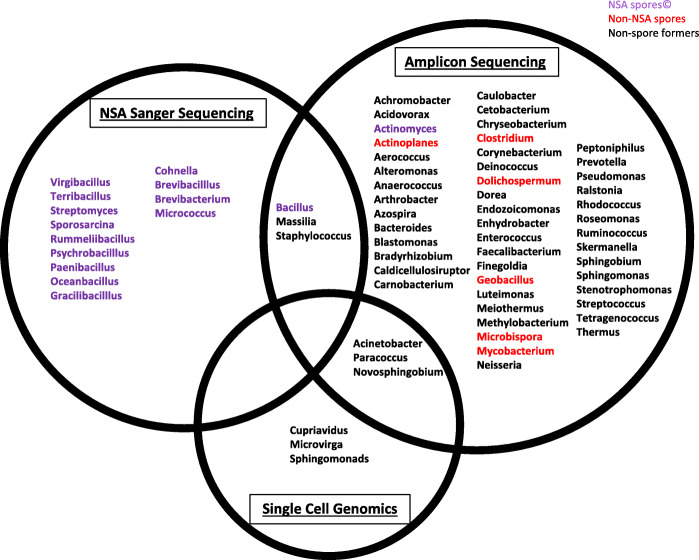
Results: Cleanroom samples were examined for total (living and dead) and viable (living only) microbial populations using molecular approaches and cultured isolates employing the traditional NASA standard spore assay (NSA), which predominantly isolated spores. The 130 NSA isolates were represented by 16 bacterial genera, of which 97% were identified as spore-formers via Sanger sequencing. The most spatially abundant isolate was Bacillus subtilis, and the most temporally abundant spore-former was Virgibacillus panthothenticus. The 16S rRNA gene-targeted amplicon sequencing detected 51 additional genera not found in the NSA method. The amplicon sequencing of the samples treated with propidium monoazide (PMA), which would differentiate between viable and dead organisms, revealed a total of 54 genera: 46 viable non-spore forming genera and 8 viable spore forming genera in these samples. The microbial diversity generated by the amplicon sequencing corresponded to ~86% non-spore-formers and ~14% spore-formers. The most common spatially distributed genera were Sphinigobium, Geobacillus, and Bacillus whereas temporally distributed common genera were Acinetobacter, Geobacilllus, and Bacillus. Single-cell genomics detected 6 genera in the sample analyzed, with the most prominent being Acinetobacter.
Conclusion: This study clearly established that detecting spores via NSA does not provide a complete assessment for the cleanliness of spacecraft-associated environments since it failed to detect several PP-relevant genera that were only recovered via molecular methods. This highlights the importance of a methodological paradigm shift to appropriately monitor bioburden in cleanrooms for not only the aeronautical industry but also for pharmaceutical, medical industries, etc., and the need to employ molecular sequencing to complement traditional culture-based assays. Video abstract.
© 2021. The Author(s).
Conflict of interest statement
All other authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
