

Time is of the essence: the molecular mechanisms of primary microcephaly
- PMID: 34862179
- PMCID: PMC8653793
- DOI: 10.1101/gad.348866.121
Time is of the essence: the molecular mechanisms of primary microcephaly
Abstract
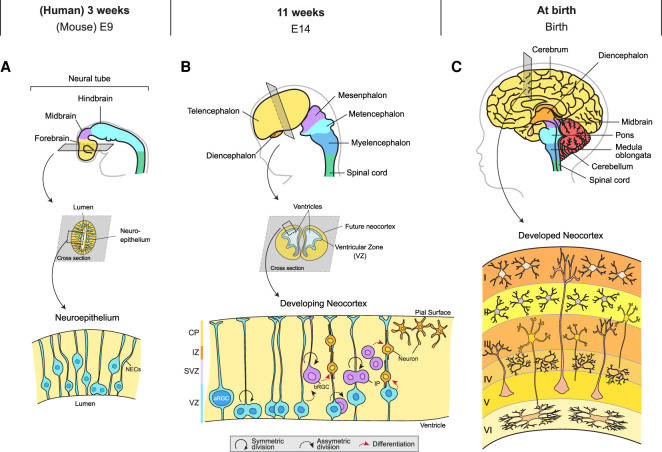
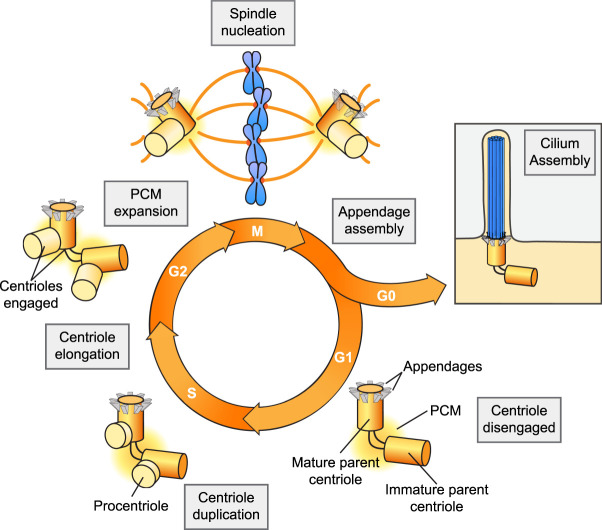
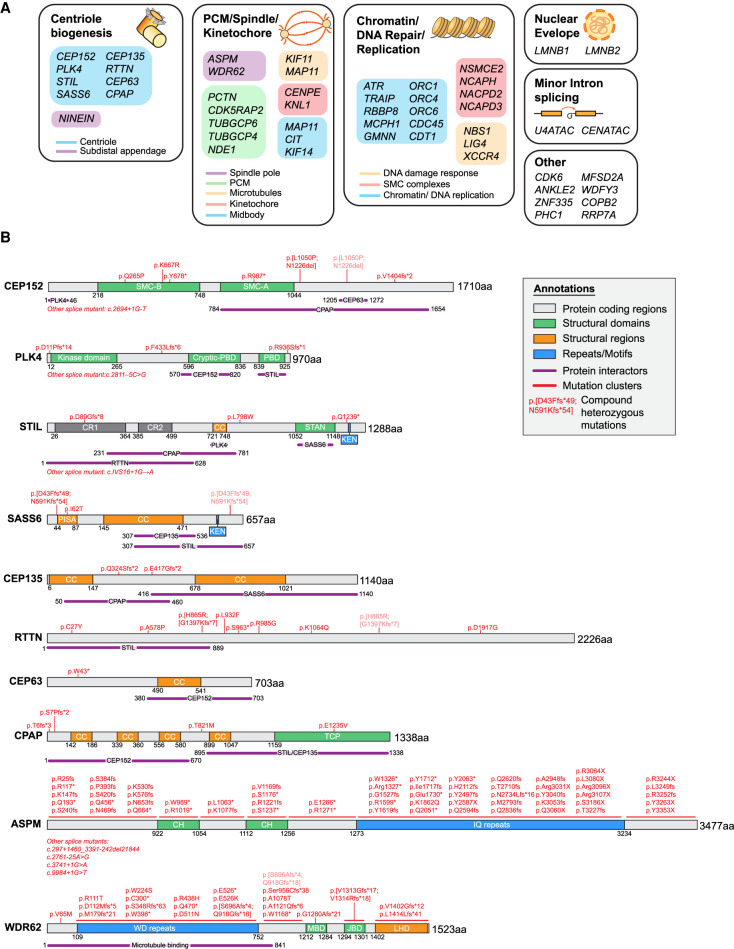
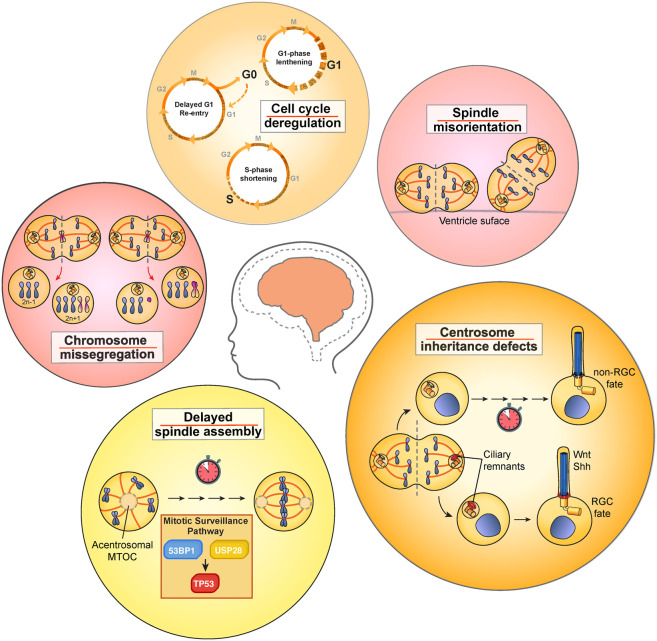
Primary microcephaly is a brain growth disorder characterized by a severe reduction of brain size and thinning of the cerebral cortex. Many primary microcephaly mutations occur in genes that encode centrosome proteins, highlighting an important role for centrosomes in cortical development. Centrosomes are microtubule organizing centers that participate in several processes, including controlling polarity, catalyzing spindle assembly in mitosis, and building primary cilia. Understanding which of these processes are altered and how these disruptions contribute to microcephaly pathogenesis is a central unresolved question. In this review, we revisit the different models that have been proposed to explain how centrosome dysfunction impairs cortical development. We review the evidence supporting a unified model in which centrosome defects reduce cell proliferation in the developing cortex by prolonging mitosis and activating a mitotic surveillance pathway. Finally, we also extend our discussion to centrosome-independent microcephaly mutations, such as those involved in DNA replication and repair.
Keywords: brain development; centriole; centrosome; cilia; microcephaly.
© 2021 Phan and Holland; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Alakbarzade V, Hameed A, Quek DQY, Chioza BA, Baple EL, Cazenave-Gassiot A, Nguyen LN, Wenk MR, Ahmad AQ, Sreekantan-Nair A, et al. 2015. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet 47: 814–817. 10.1038/ng.3313 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources