

Most myopathic lamin variants aggregate: a functional genomics approach for assessing variants of uncertain significance
- PMID: 34862408
- PMCID: PMC8642518
- DOI: 10.1038/s41525-021-00265-x
Most myopathic lamin variants aggregate: a functional genomics approach for assessing variants of uncertain significance
Abstract
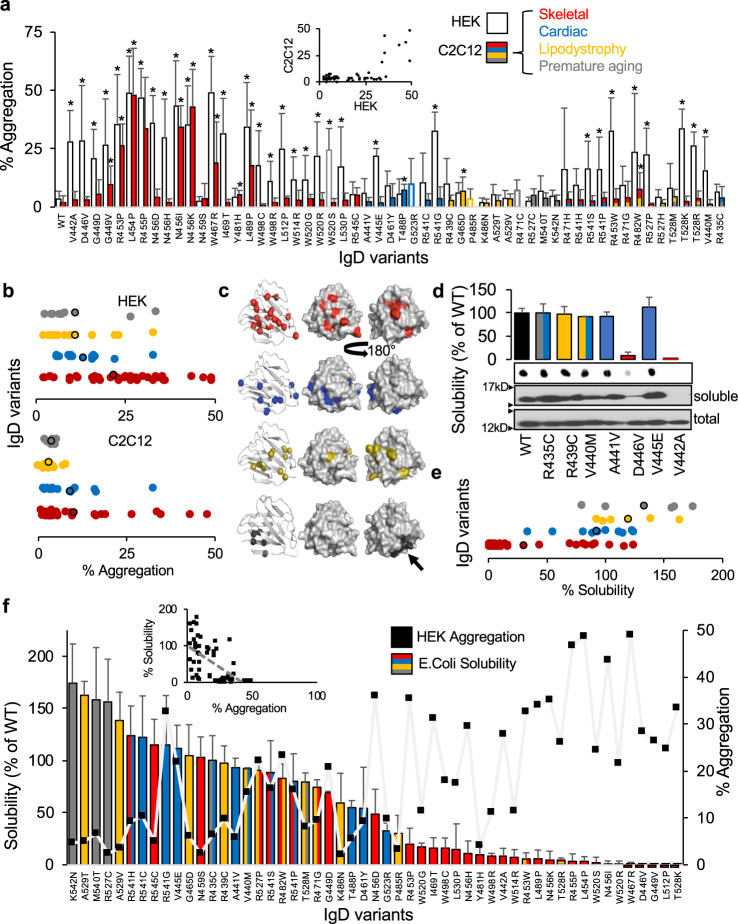
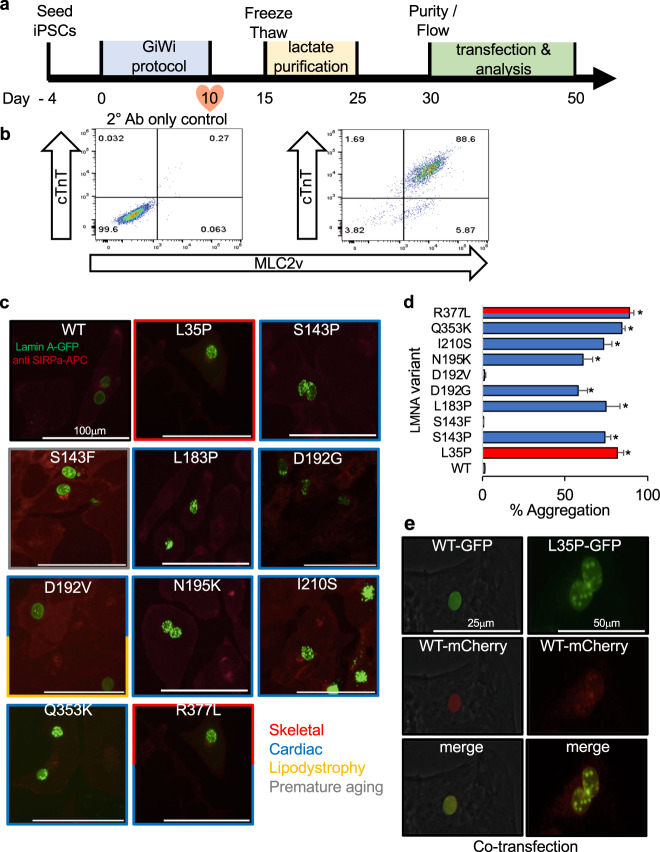
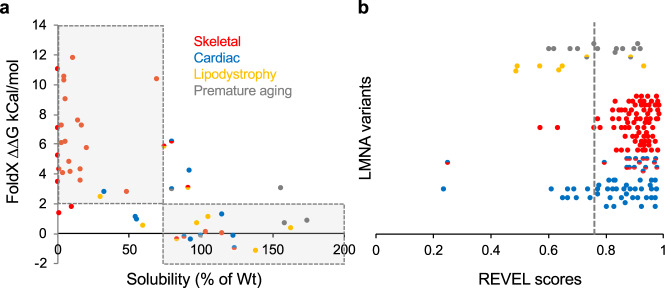
Hundreds of LMNA variants have been associated with several distinct disease phenotypes. However, genotype-phenotype relationships remain largely undefined and the impact for most variants remains unknown. We performed a functional analysis for 178 variants across five structural domains using two different overexpression models. We found that lamin A aggregation is a major determinant for skeletal and cardiac laminopathies. An in vitro solubility assay shows that aggregation-prone variants in the immunoglobulin-like domain correlate with domain destabilization. Finally, we demonstrate that myopathic-associated LMNA variants show aggregation patterns in induced pluripotent stem cell derived-cardiomyocytes (iPSC-CMs) in contrast to non-myopathic LMNA variants. Our data-driven approach (1) reveals that striated muscle laminopathies are predominantly protein misfolding diseases, (2) demonstrates an iPSC-CM experimental platform for characterizing laminopathic variants in human cardiomyocytes, and (3) supports a functional assay to aid in assessing pathogenicity for myopathic variants of uncertain significance.
© 2021. The Author(s).
Conflict of interest statement
T.J.K. serves as a consultant for Fujifilm Cellular Dynamics, which is a stem cell technology company. The other authors declare no other conflict of interests or financial disclosures to declare.
Figures



References
-
- Bonne G, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999;21:285–288. - PubMed
-
- Fatkin D, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999;341:1715–1724. - PubMed
-
- Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000;9:109–112. - PubMed
Grants and funding
- R01 HL139738/HL/NHLBI NIH HHS/United States
- R01 HL128598/HL/NHLBI NIH HHS/United States
- R01 HL139738-01/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- F32 HL128091/HL/NHLBI NIH HHS/United States
- R01 HL07887/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
