Insight into the molecular basis of substrate recognition by the wall teichoic acid glycosyltransferase TagA
- PMID: 34864059
- PMCID: PMC8784642
- DOI: 10.1016/j.jbc.2021.101464
Insight into the molecular basis of substrate recognition by the wall teichoic acid glycosyltransferase TagA
Abstract
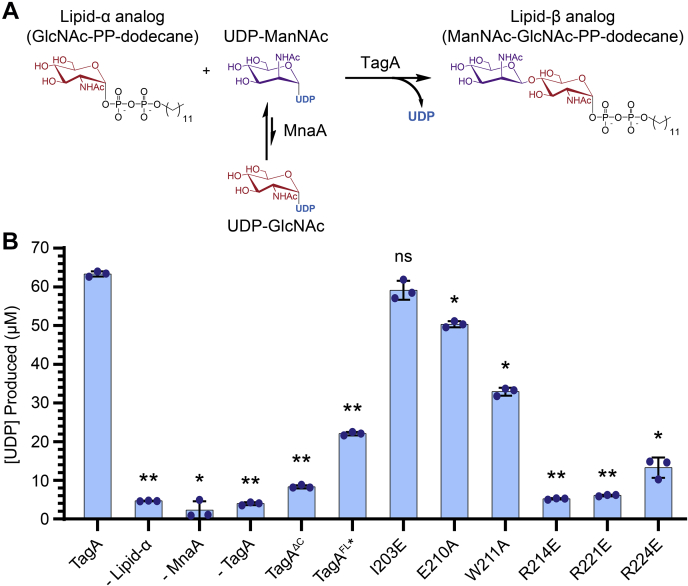
Wall teichoic acid (WTA) polymers are covalently affixed to the Gram-positive bacterial cell wall and have important functions in cell elongation, cell morphology, biofilm formation, and β-lactam antibiotic resistance. The first committed step in WTA biosynthesis is catalyzed by the TagA glycosyltransferase (also called TarA), a peripheral membrane protein that produces the conserved linkage unit, which joins WTA to the cell wall peptidoglycan. TagA contains a conserved GT26 core domain followed by a C-terminal polypeptide tail that is important for catalysis and membrane binding. Here, we report the crystal structure of the Thermoanaerobacter italicus TagA enzyme bound to UDP-N-acetyl-d-mannosamine, revealing the molecular basis of substrate binding. Native MS experiments support the model that only monomeric TagA is enzymatically active and that it is stabilized by membrane binding. Molecular dynamics simulations and enzyme activity measurements indicate that the C-terminal polypeptide tail facilitates catalysis by encapsulating the UDP-N-acetyl-d-mannosamine substrate, presenting three highly conserved arginine residues to the active site that are important for catalysis (R214, R221, and R224). From these data, we present a mechanistic model of catalysis that ascribes functions for these residues. This work could facilitate the development of new antimicrobial compounds that disrupt WTA biosynthesis in pathogenic bacteria.
Keywords: MS; NMR; TagA; Thermoanaerobacter italicus; X-ray crystallography; glycosyltransferase; methicillin-resistant Staphylococcus aureus; molecular dynamics; peripheral membrane protein; teichoic acid.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures