

The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance
- PMID: 34867178
- PMCID: PMC8634724
- DOI: 10.3389/fnins.2021.784987
The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance
Abstract
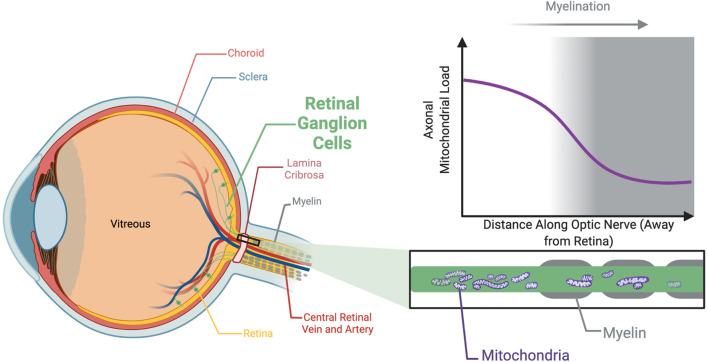
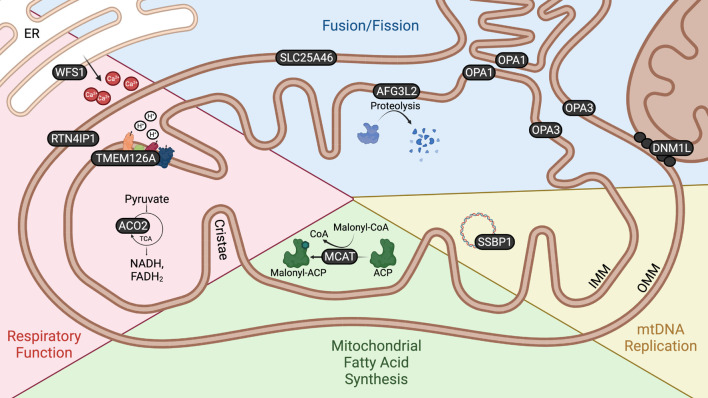
Optic atrophy (OA) with autosomal inheritance is a form of optic neuropathy characterized by the progressive and irreversible loss of vision. In some cases, this is accompanied by additional, typically neurological, extra-ocular symptoms. Underlying the loss of vision is the specific degeneration of the retinal ganglion cells (RGCs) which form the optic nerve. Whilst autosomal OA is genetically heterogenous, all currently identified causative genes appear to be associated with mitochondrial organization and function. However, it is unclear why RGCs are particularly vulnerable to mitochondrial aberration. Despite the relatively high prevalence of this disorder, there are currently no approved treatments. Combined with the lack of knowledge concerning the mechanisms through which aberrant mitochondrial function leads to RGC death, there remains a clear need for further research to identify the underlying mechanisms and develop treatments for this condition. This review summarizes the genes known to be causative of autosomal OA and the mitochondrial dysfunction caused by pathogenic mutations. Furthermore, we discuss the suitability of available in vivo models for autosomal OA with regards to both treatment development and furthering the understanding of autosomal OA pathology.
Keywords: in vivo models; mitochondria; optic atrophy; retinal ganglion cells (RGC); retinal organoids.
Copyright © 2021 Strachan, Mac White-Begg, Crean, Reynolds, Kennedy and O’Sullivan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ali M. S., Suda K., Kowada R., Ueoka I., Yoshida H., Yamaguchi M. (2020). Neuron-specific knockdown of solute carrier protein SLC25A46a induces locomotive defects, an abnormal neuron terminal morphology, learning disability, and shortened lifespan. IBRO Rep. 8 65–75. 10.1016/j.ibror.2020.02.001 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases