

Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome
- PMID: 34867935
- PMCID: PMC8634673
- DOI: 10.3389/fimmu.2021.656797
Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome
Abstract
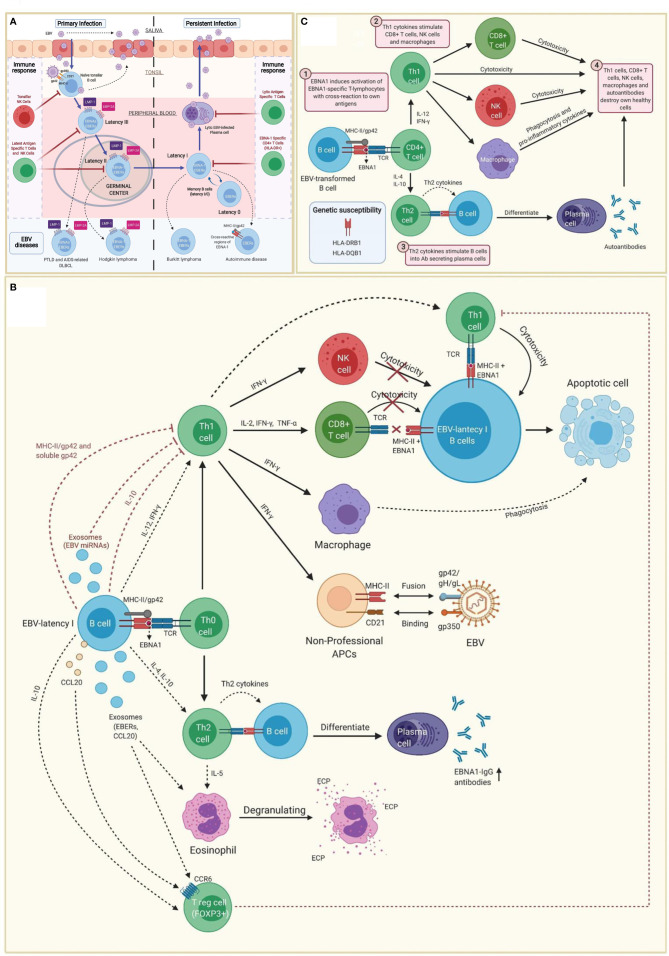
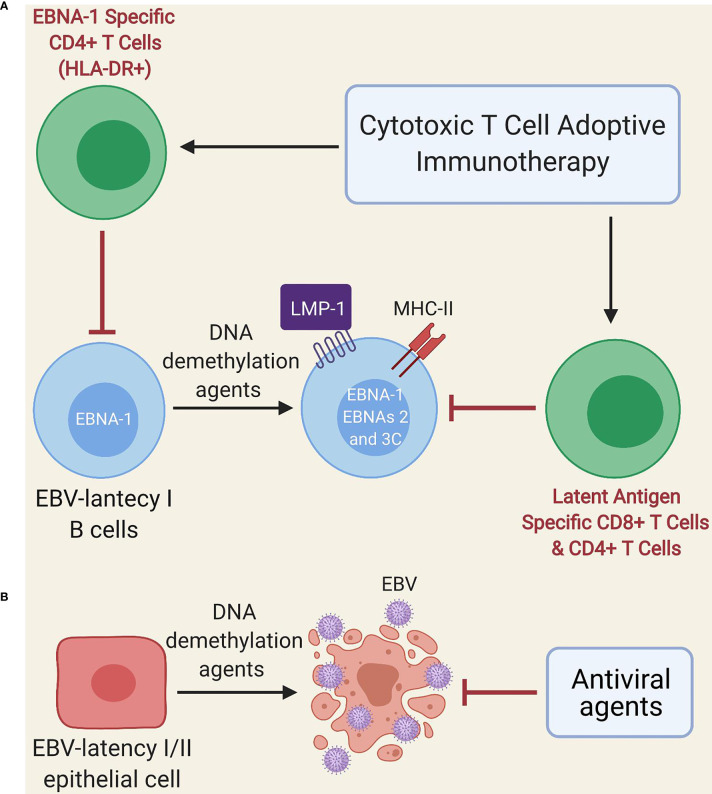
Myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS) affects approximately 1% of the general population. It is a chronic, disabling, multi-system disease for which there is no effective treatment. This is probably related to the limited knowledge about its origin. Here, we summarized the current knowledge about the pathogenesis of ME/CFS and revisit the immunopathobiology of Epstein-Barr virus (EBV) infection. Given the similarities between EBV-associated autoimmune diseases and cancer in terms of poor T cell surveillance of cells with EBV latency, expanded EBV-infected cells in peripheral blood and increased antibodies against EBV, we hypothesize that there could be a common etiology generated by cells with EBV latency that escape immune surveillance. Albeit inconclusive, multiple studies in patients with ME/CFS have suggested an altered cellular immunity and augmented Th2 response that could result from mechanisms of evasion to some pathogens such as EBV, which has been identified as a risk factor in a subset of ME/CFS patients. Namely, cells with latency may evade the immune system in individuals with genetic predisposition to develop ME/CFS and in consequence, there could be poor CD4 T cell immunity to mitogens and other specific antigens, as it has been described in some individuals. Ultimately, we hypothesize that within ME/CFS there is a subgroup of patients with DRB1 and DQB1 alleles that could confer greater susceptibility to EBV, where immune evasion mechanisms generated by cells with latency induce immunodeficiency. Accordingly, we propose new endeavors to investigate if anti-EBV therapies could be effective in selected ME/CFS patients.
Keywords: CD4+ CTL; EBV EBNA-1; HLA-II alleles; autoimmunity; cancer; chronic fatigue syndrome; immunotherapy; myalgic encephalomyelitis.
Copyright © 2021 Ruiz-Pablos, Paiva, Montero-Mateo, Garcia and Zabaleta.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials