

Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function
- PMID: 34869339
- PMCID: PMC8639223
- DOI: 10.3389/fcell.2021.751590
Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function
Abstract
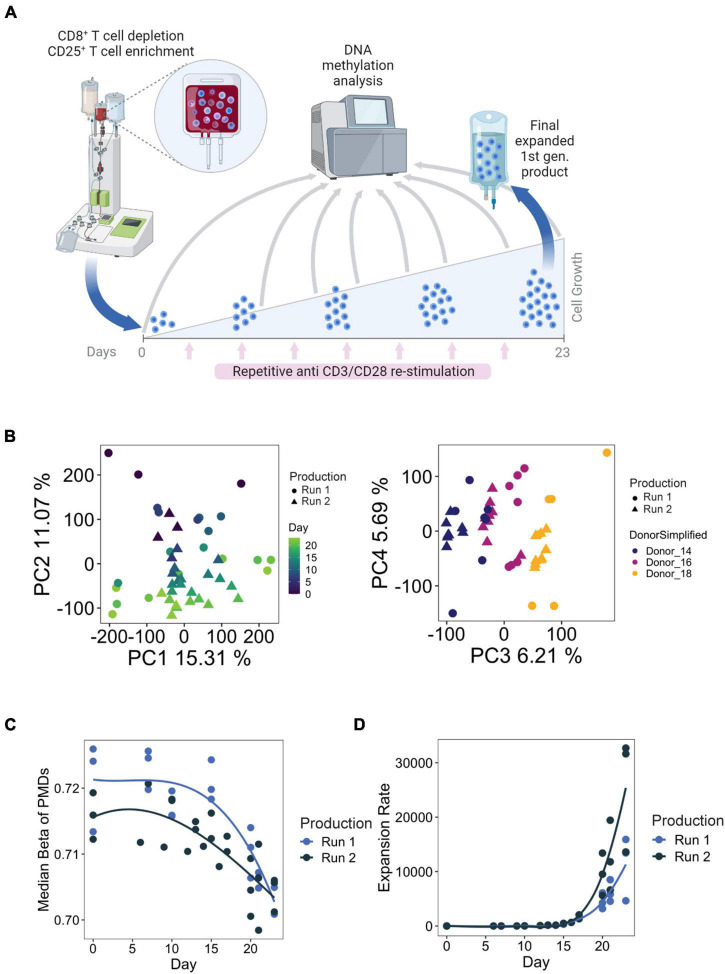
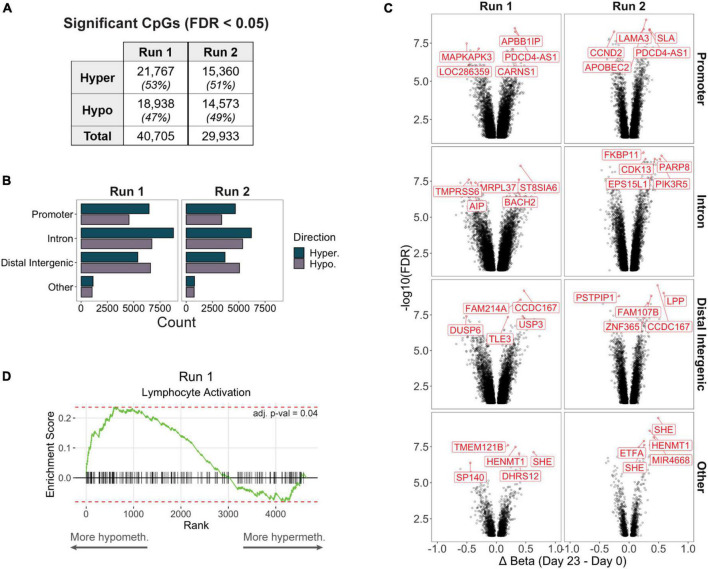
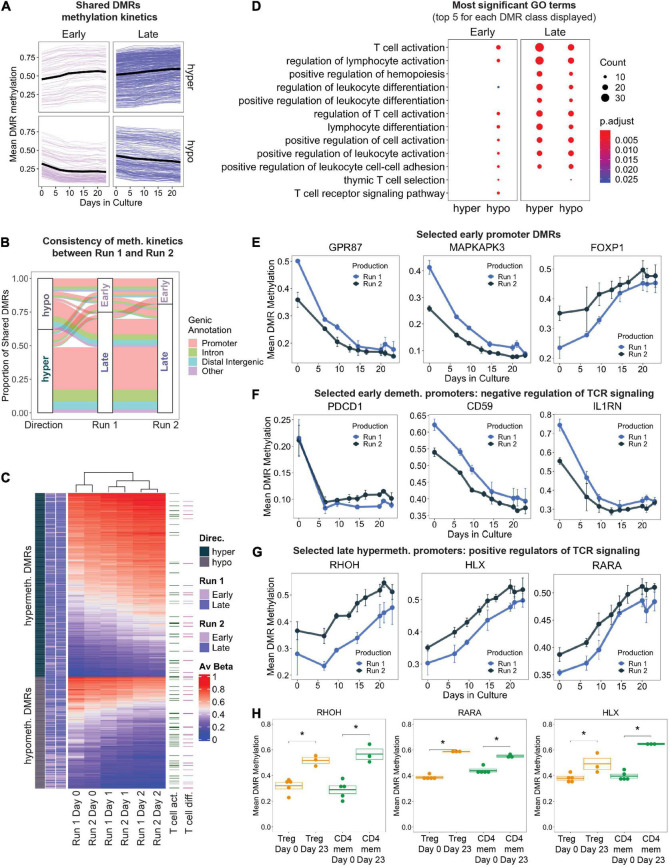
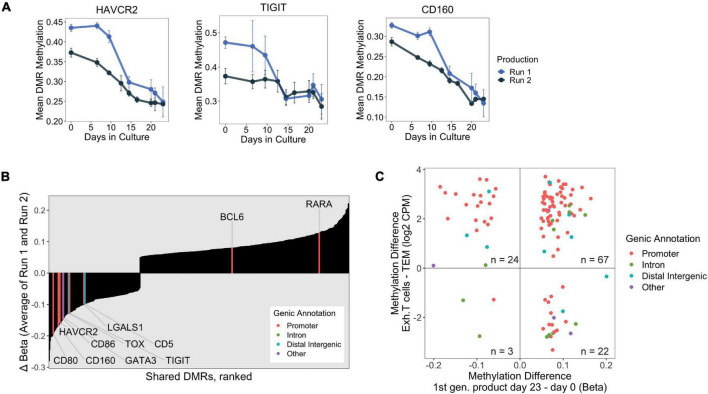
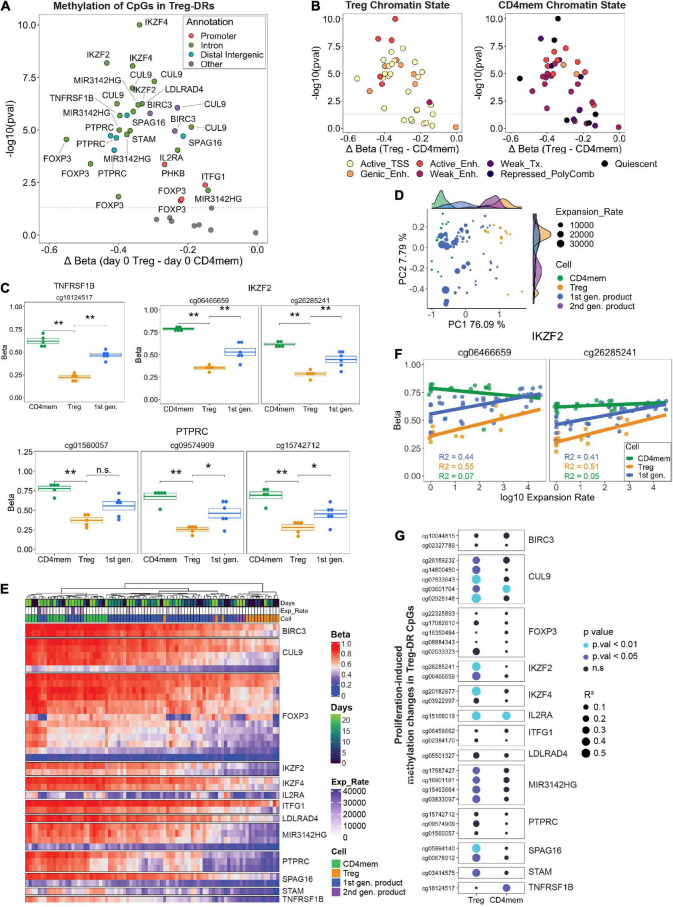
Adoptive transfer of regulatory T cells (Treg) is a promising new therapeutic option to treat detrimental inflammatory conditions after transplantation and during autoimmune disease. To reach sufficient cell yield for treatment, ex vivo isolated autologous or allogenic Tregs need to be expanded extensively in vitro during manufacturing of the Treg product. However, repetitive cycles of restimulation and prolonged culture have been shown to impact T cell phenotypes, functionality and fitness. It is therefore critical to scrutinize the molecular changes which occur during T cell product generation, and reexamine current manufacturing practices. We performed genome-wide DNA methylation profiling of cells throughout the manufacturing process of a polyclonal Treg product that has proven safety and hints of therapeutic efficacy in kidney transplant patients. We found progressive DNA methylation changes over the duration of culture, which were donor-independent and reproducible between manufacturing runs. Differentially methylated regions (DMRs) in the final products were significantly enriched at promoters and enhancers of genes implicated in T cell activation. Additionally, significant hypomethylation did also occur in promoters of genes implicated in functional exhaustion in conventional T cells, some of which, however, have been reported to strengthen immunosuppressive effector function in Tregs. At the same time, a set of reported Treg-specific demethylated regions increased methylation levels with culture, indicating a possible destabilization of Treg identity during manufacturing, which was independent of the purity of the starting material. Together, our results indicate that the repetitive TCR-mediated stimulation lead to epigenetic changes that might impact functionality of Treg products in multiple ways, by possibly shifting to an effector Treg phenotype with enhanced functional activity or by risking destabilization of Treg identity and impaired TCR activation. Our analyses also illustrate the value of epigenetic profiling for the evaluation of T cell product manufacturing pipelines, which might open new avenues for the improvement of current adoptive Treg therapies with relevance for conventional effector T cell products.
Keywords: DNA methylation; adoptive cell therapy; advanced therapy medicinal products; biomarker; good manufacturing practice; regulatory T cells.
Copyright © 2021 Ou, Hamo, Schulze, Roemhild, Kaiser, Gasparoni, Salhab, Zarrinrad, Amini, Schlickeiser, Streitz, Walter, Volk, Schmueck-Henneresse, Reinke and Polansky.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Allen J. D., Harris A. W., Bath M. L., Strasser A., Scollay R., Adams J. M. (1995). Perturbed development of T and B cells in mice expressing an Hlx homeobox transgene. J. Immunol. 154 1531–1542. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources