

Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms
- PMID: 34870237
- PMCID: PMC7612062
- DOI: 10.1038/s43018-021-00259-9
Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms
Abstract
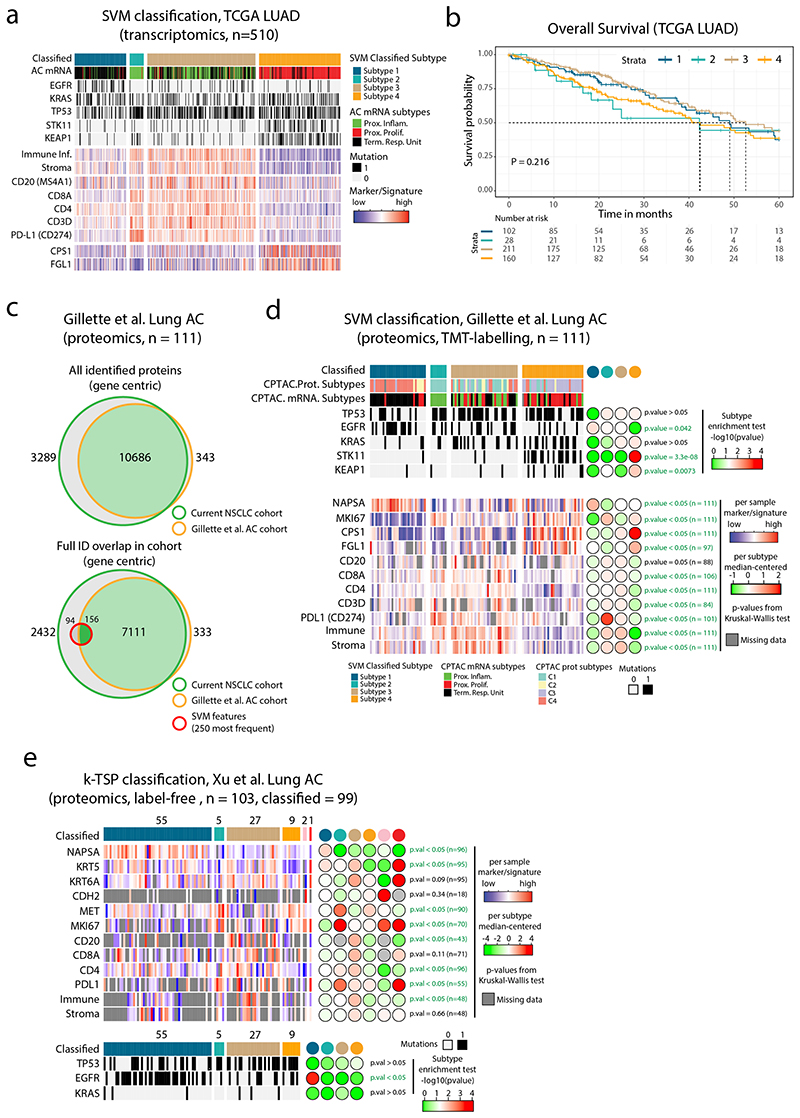
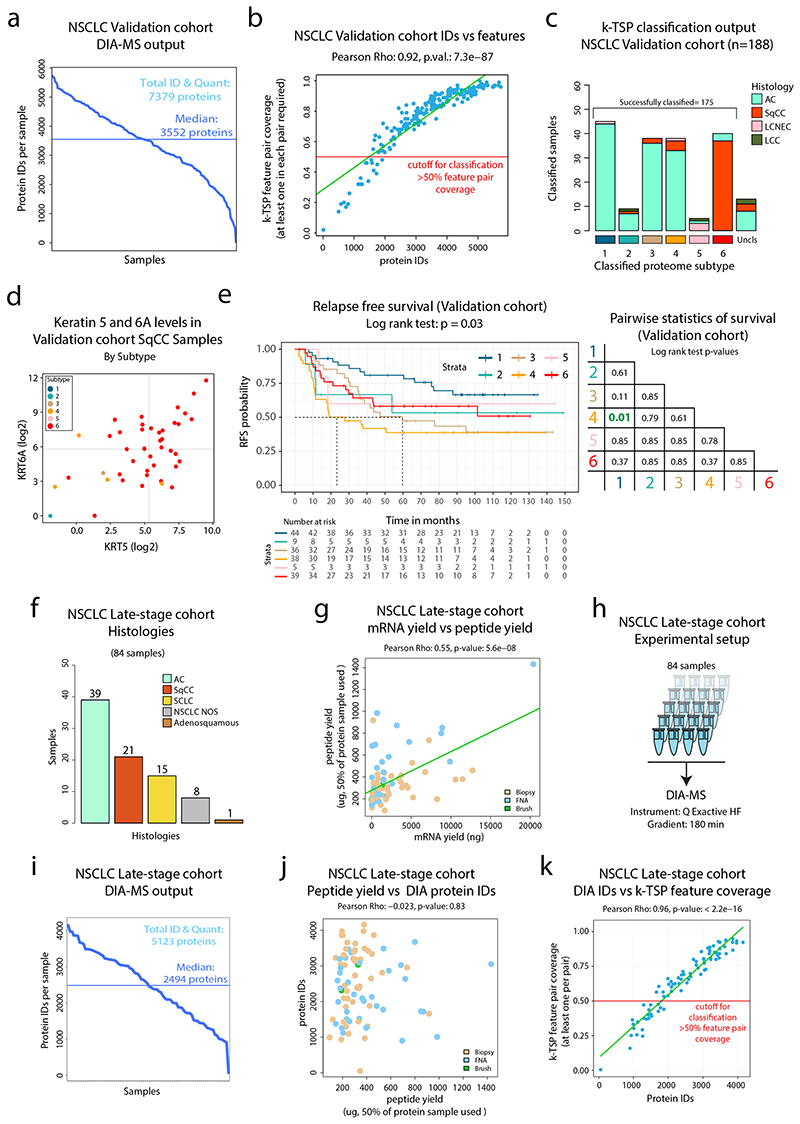
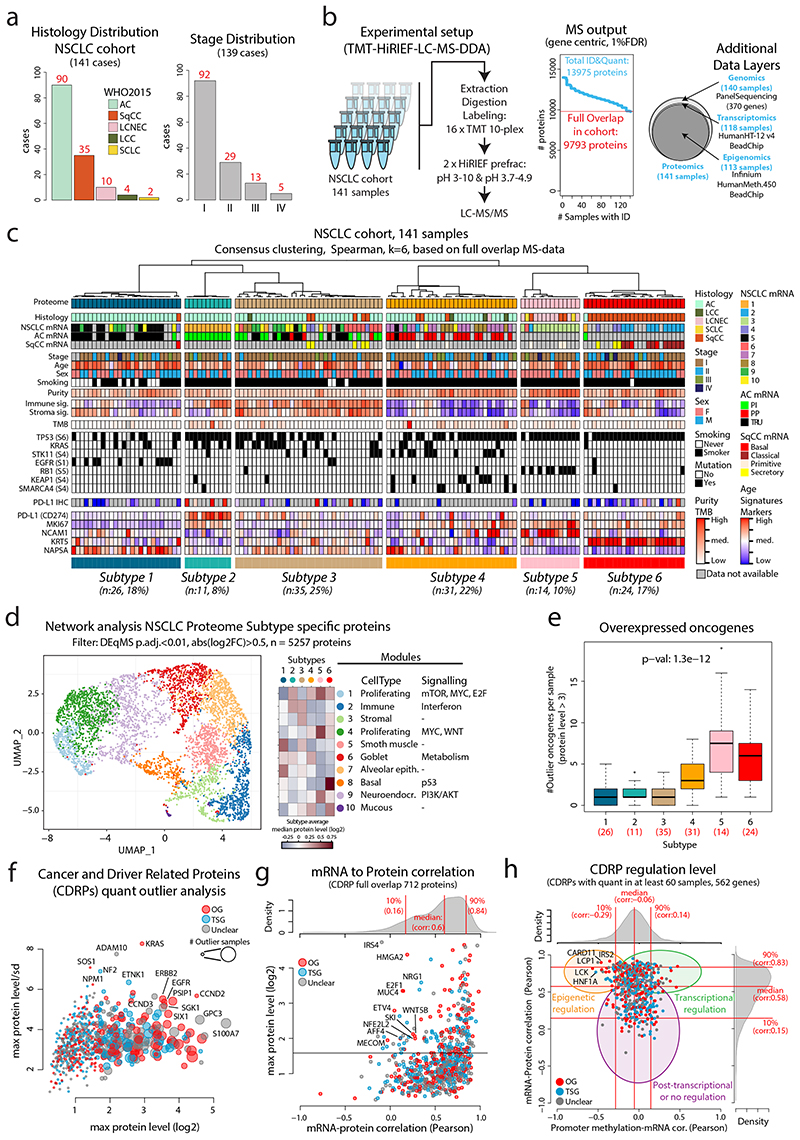
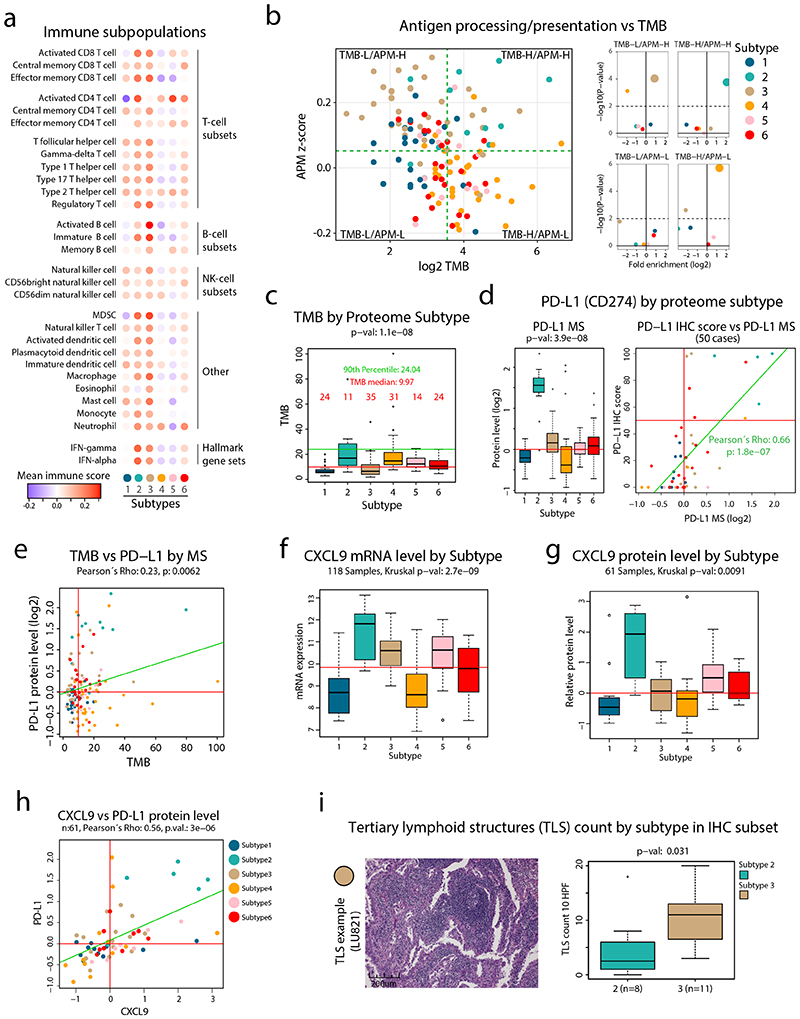
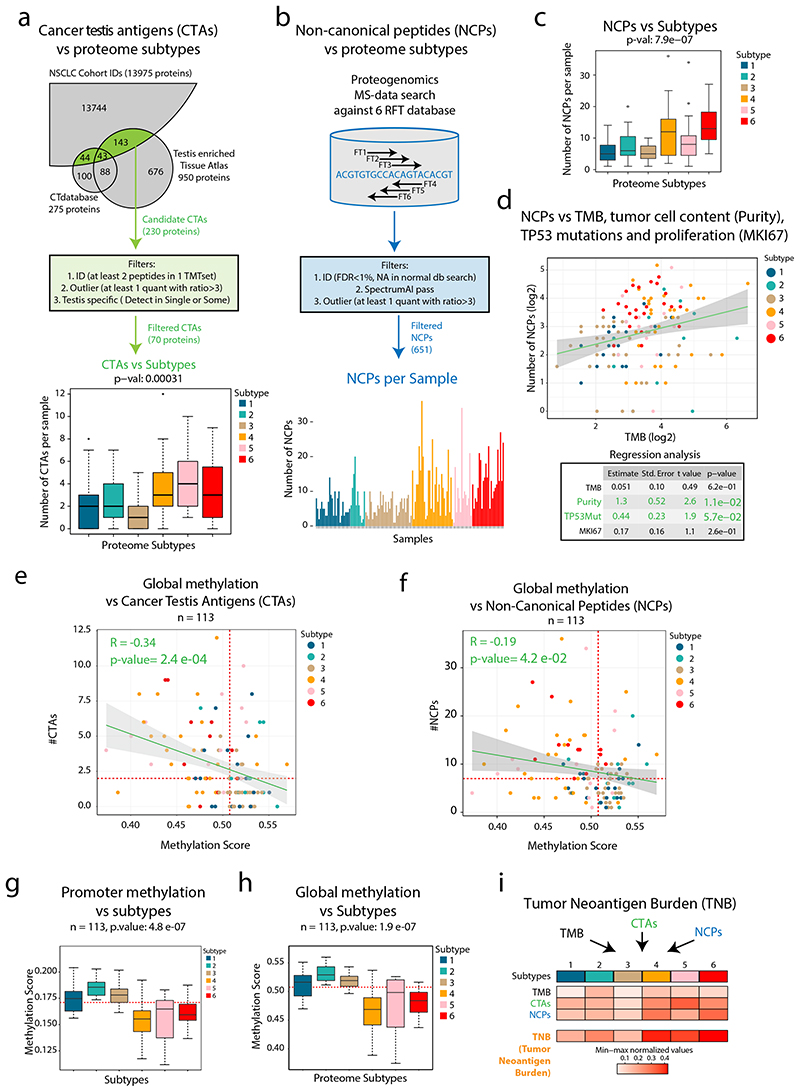
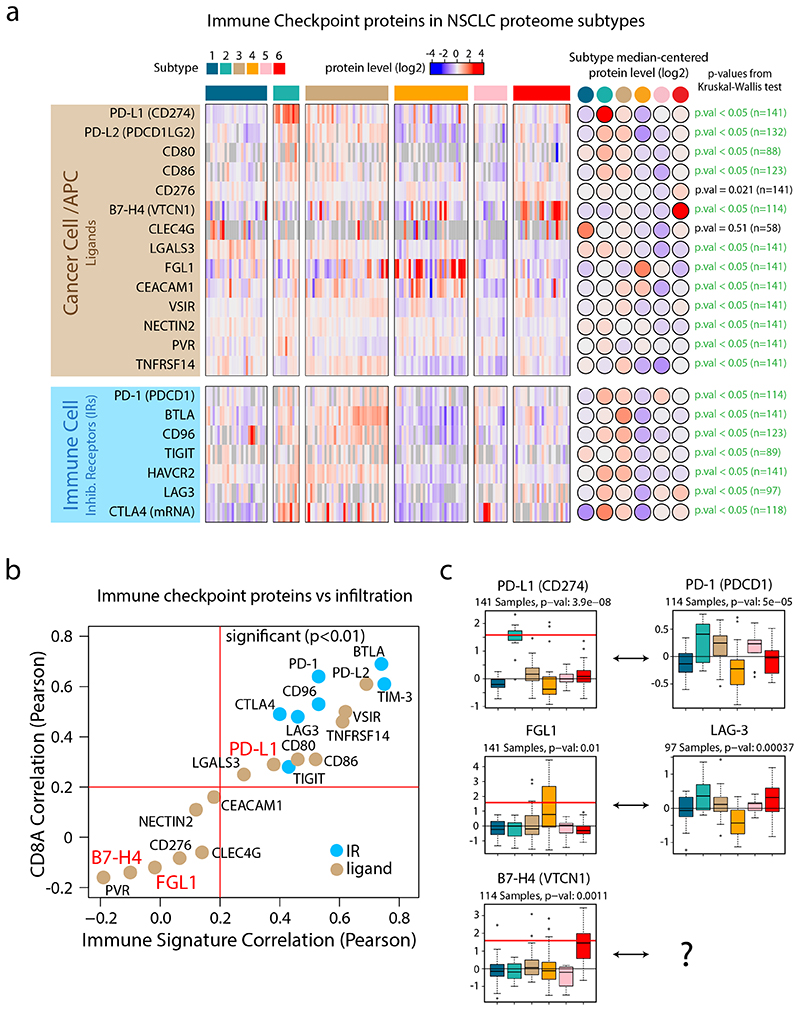
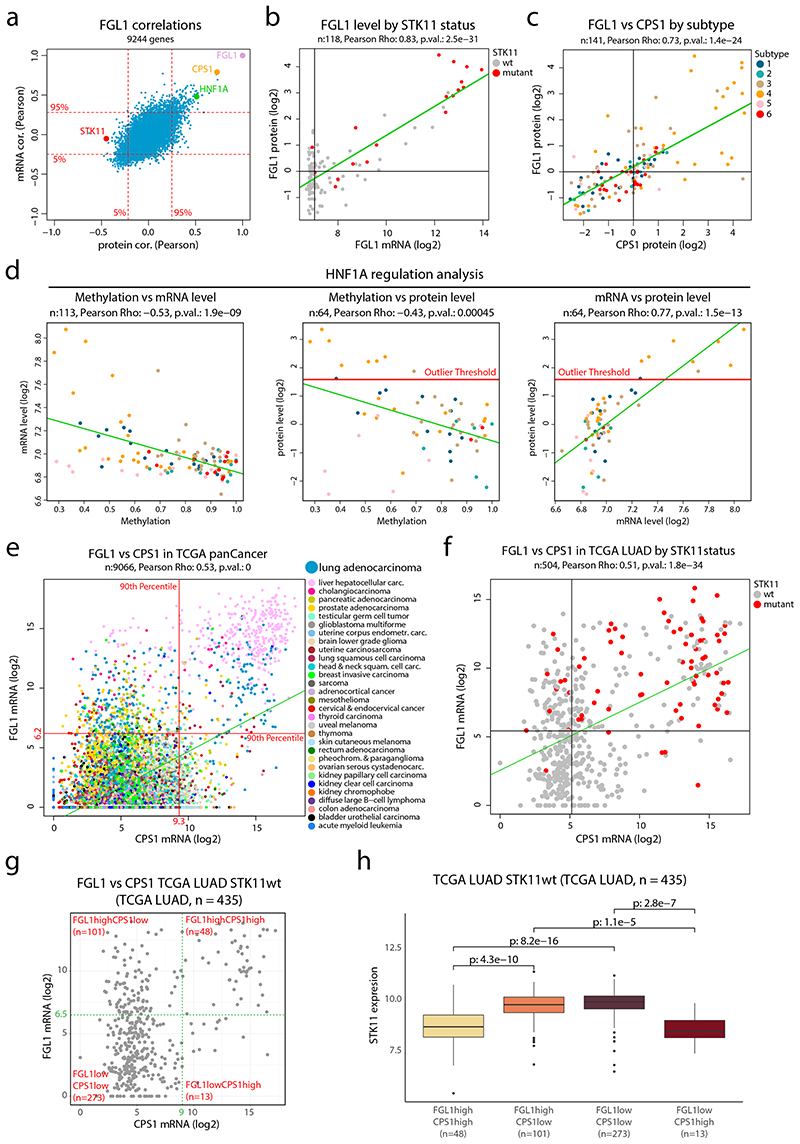
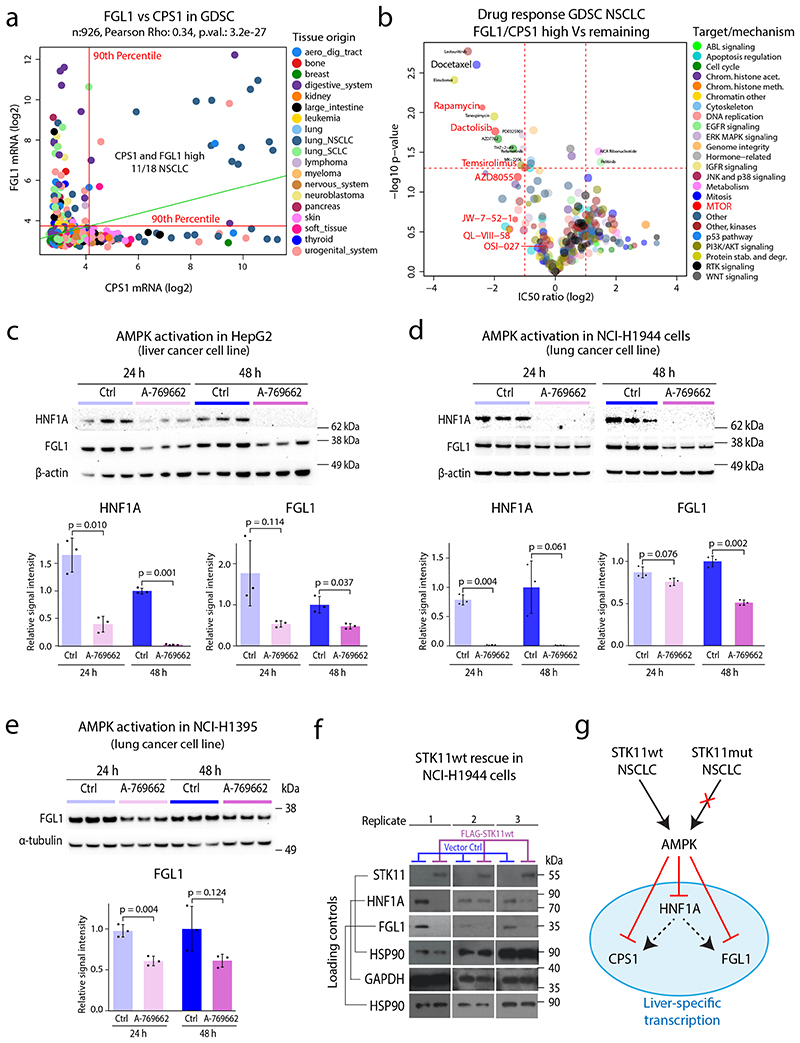
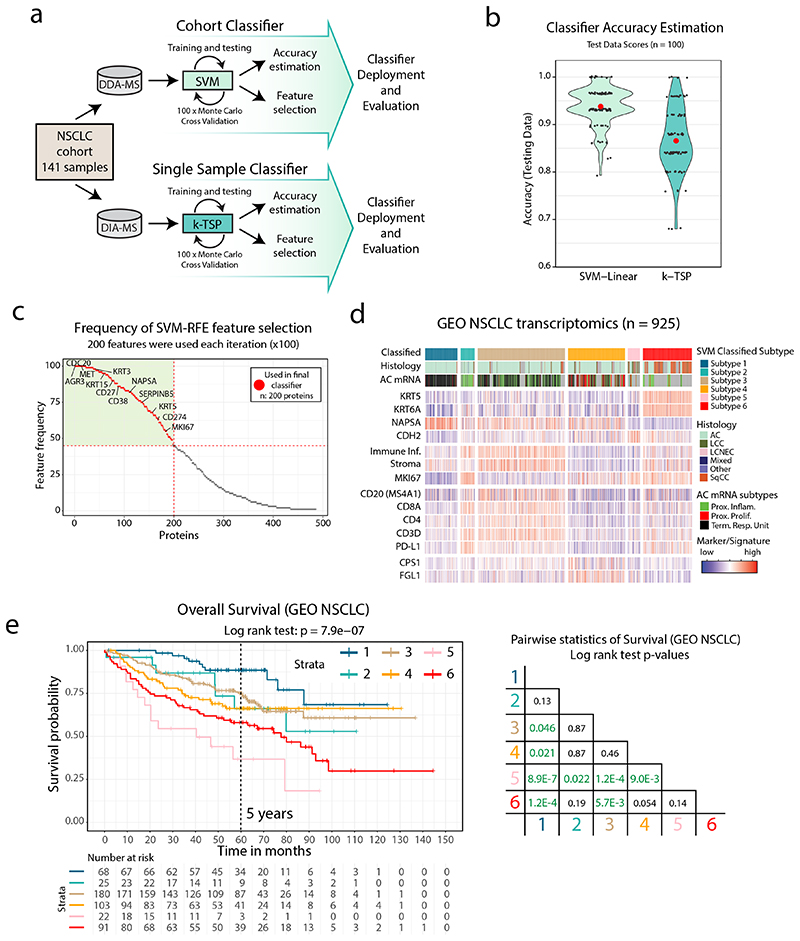
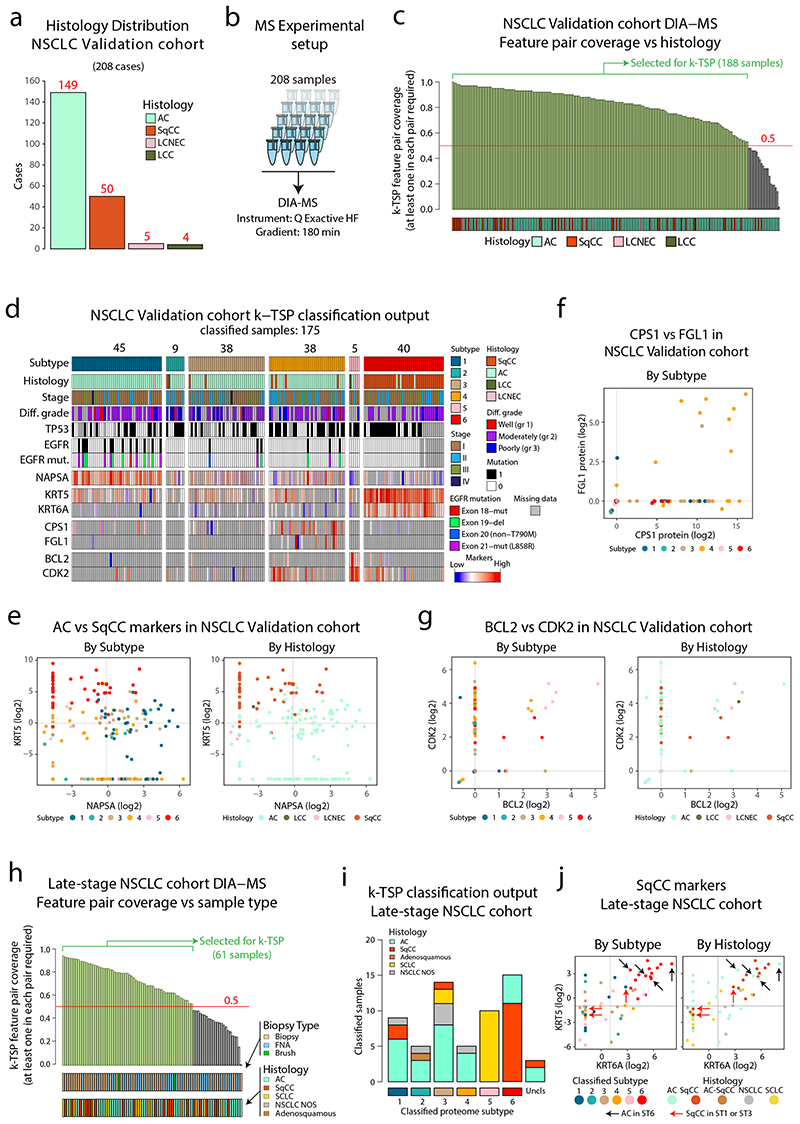
Despite major advancements in lung cancer treatment, long-term survival is still rare, and a deeper understanding of molecular phenotypes would allow the identification of specific cancer dependencies and immune evasion mechanisms. Here we performed in-depth mass spectrometry (MS)-based proteogenomic analysis of 141 tumors representing all major histologies of non-small cell lung cancer (NSCLC). We identified six distinct proteome subtypes with striking differences in immune cell composition and subtype-specific expression of immune checkpoints. Unexpectedly, high neoantigen burden was linked to global hypomethylation and complex neoantigens mapped to genomic regions, such as endogenous retroviral elements and introns, in immune-cold subtypes. Further, we linked immune evasion with LAG3 via STK11 mutation-dependent HNF1A activation and FGL1 expression. Finally, we develop a data-independent acquisition MS-based NSCLC subtype classification method, validate it in an independent cohort of 208 NSCLC cases and demonstrate its clinical utility by analyzing an additional cohort of 84 late-stage NSCLC biopsy samples.
Conflict of interest statement
Competing interests J.L. has received grant funding from AstraZeneca, Roche and Novartis (not financing of the current manuscript). J.L. and L.M.O. are share holders of FenoMark Diagnostics. J.L., T.A., I.S., and L.M.O are co-inventors on a patent application related to this work. J.L. and D.T. are associate with Roche financed Cancer Core Europe clinical trial (not associated to current manuscript). Since completing his contribution to the current work, M.Pirmoradian has become an employee of AstraZeneca. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
