

Sequencing-based genome-wide association studies reporting standards
- PMID: 34870259
- PMCID: PMC8637874
- DOI: 10.1016/j.xgen.2021.100005
Sequencing-based genome-wide association studies reporting standards
Abstract
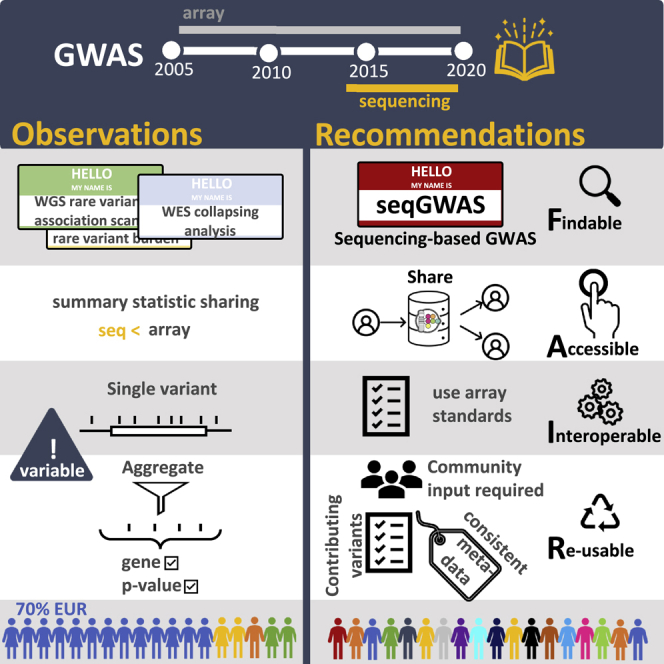
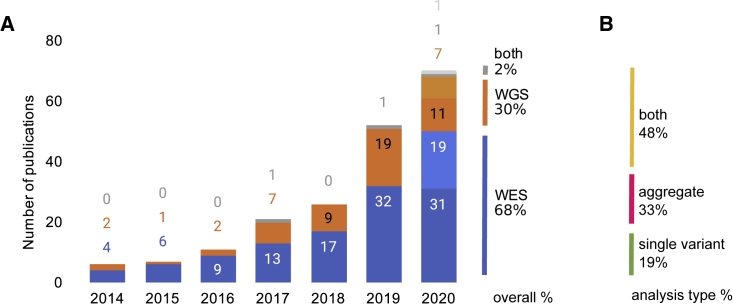
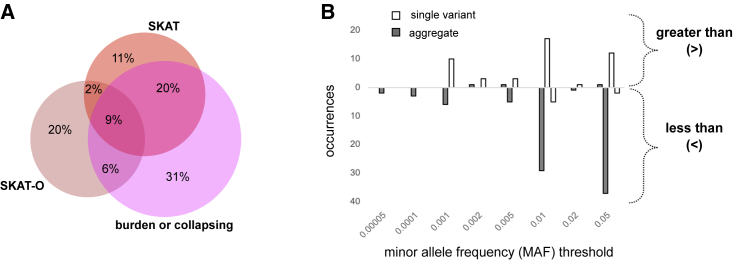
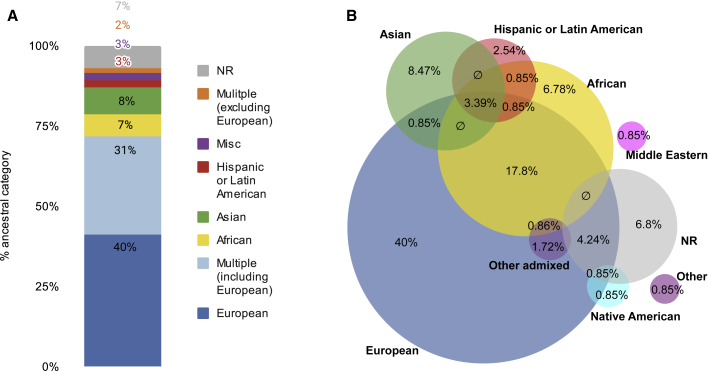
Genome sequencing has recently become a viable genotyping technology for use in genome-wide association studies (GWASs), offering the potential to analyze a broader range of genome-wide variation, including rare variants. To survey current standards, we assessed the content and quality of reporting of statistical methods, analyses, results, and datasets in 167 exome- or genome-wide-sequencing-based GWAS publications published from 2014 to 2020; 81% of publications included tests of aggregate association across multiple variants, with multiple test models frequently used. We observed a lack of standardized terms and incomplete reporting of datasets, particularly for variants analyzed in aggregate tests. We also find a lower frequency of sharing of summary statistics compared with array-based GWASs. Reporting standards and increased data sharing are required to ensure sequencing-based association study data are findable, interoperable, accessible, and reusable (FAIR). To support that, we recommend adopting the standard terminology of sequencing-based GWAS (seqGWAS). Further, we recommend that single-variant analyses be reported following the same standards and conventions as standard array-based GWASs and be shared in the GWAS Catalog. We also provide initial recommended standards for aggregate analyses metadata and summary statistics.
Conflict of interest statement
An immediate family member of J.A.L.M. is an employee and shareholder of Illumina.
Figures
References
-
- Klein R.J., Xu X., Mukherjee S., Willis J., Hayes J. Successes of genome-wide association studies. Cell. 2010;142:350–351. author reply 353–355. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
