

SARS-CoV-2 genetic variations associated with COVID-19 pathogenicity
- PMID: 34870573
- PMCID: PMC8767342
- DOI: 10.1099/mgen.0.000734
SARS-CoV-2 genetic variations associated with COVID-19 pathogenicity
Abstract
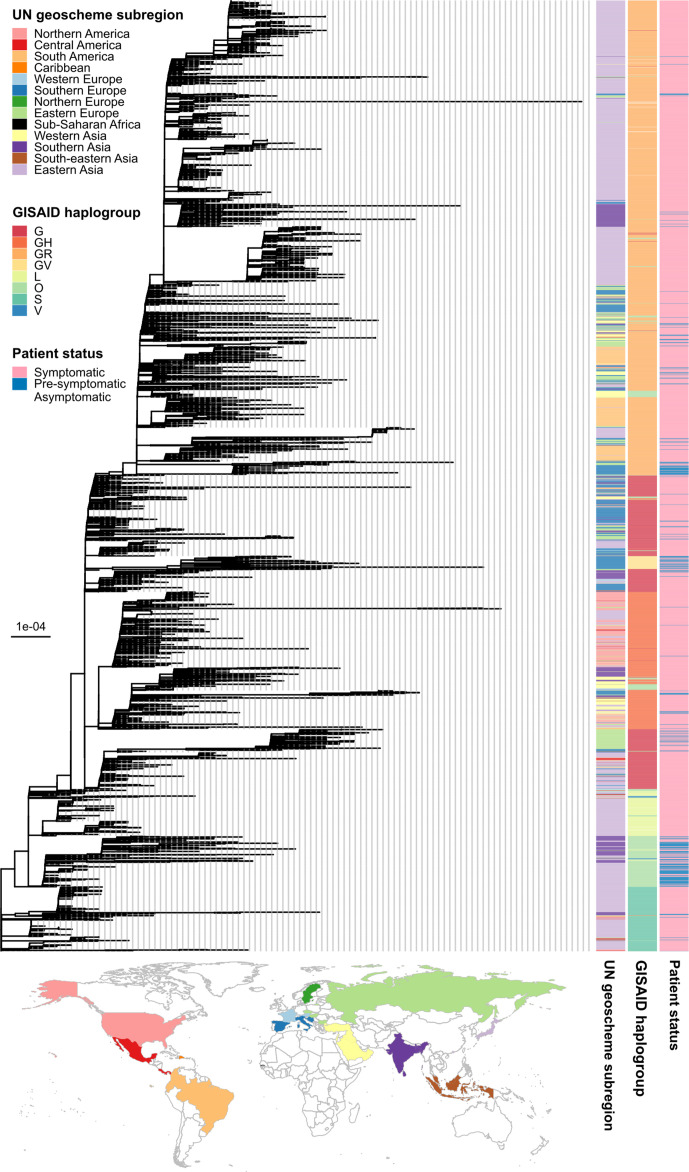
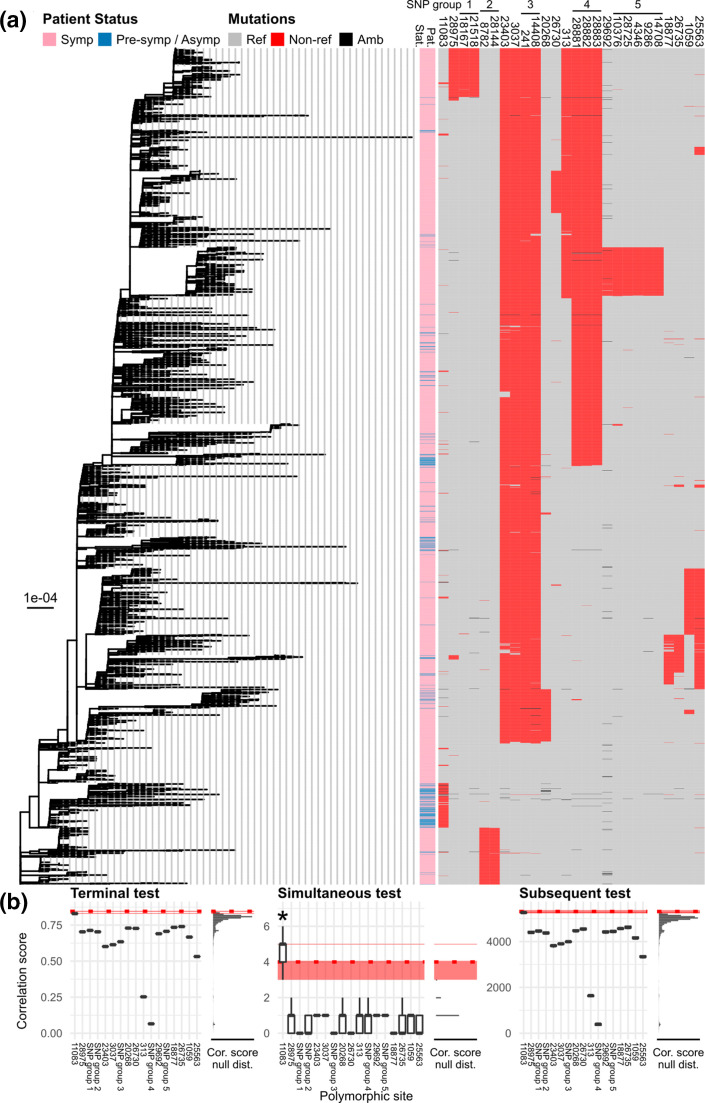
In this study, we performed genome-wide association analyses on SARS-CoV-2 genomes to identify genetic mutations associated with pre-symptomatic/asymptomatic COVID-19 cases. Various potential covariates and confounding factors of COVID-19 severity, including patient age, gender and country, as well as virus phylogenetic relatedness were adjusted for. In total, 3021 full-length genomes of SARS-CoV-2 generated from original clinical samples and whose patient status could be determined conclusively as either 'pre-symptomatic/asymptomatic' or 'symptomatic' were retrieved from the GISAID database. We found that the mutation 11 083G>T, located in the coding region of non-structural protein 6, is significantly associated with asymptomatic COVID-19. Patient age is positively correlated with symptomatic infection, while gender is not significantly correlated with the development of the disease. We also found that the effects of the mutation, patient age and gender do not vary significantly among countries, although each country appears to have varying baseline chances of COVID-19 symptom development.
Keywords: GWAS; SARS-CoV-2; nsp6.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous