

Expanding the phenotypic spectrum of mutations in LRP2: a novel candidate gene of non-syndromic familial comitant strabismus
- PMID: 34872573
- PMCID: PMC8647414
- DOI: 10.1186/s12967-021-03155-z
Expanding the phenotypic spectrum of mutations in LRP2: a novel candidate gene of non-syndromic familial comitant strabismus
Abstract
Background: Comitant strabismus (CS) is a heterogeneous disorder that is a major contributing factor to unilateral childhood-onset visual impairment. Studies have confirmed that genetic factors play an important role in the development of CS. The aim of this study was to identify the genetic cause of non-syndromic familial CS.
Methods: Fourteen unrelated CS families were recruited for the study. Twelve affected and 2 unaffected individuals from a large four-generation family (CS08) were selected to perform whole genome-wide linkage analysis. Parallel whole-exome sequencing (WES) was conducted in the same family (9 patients and 1 unaffected member) and 31 additional CS cases from 13 other unrelated families. Sanger sequencing was used to determine whether any of the remaining variants co-segregated with the disease phenotype in the corresponding family.
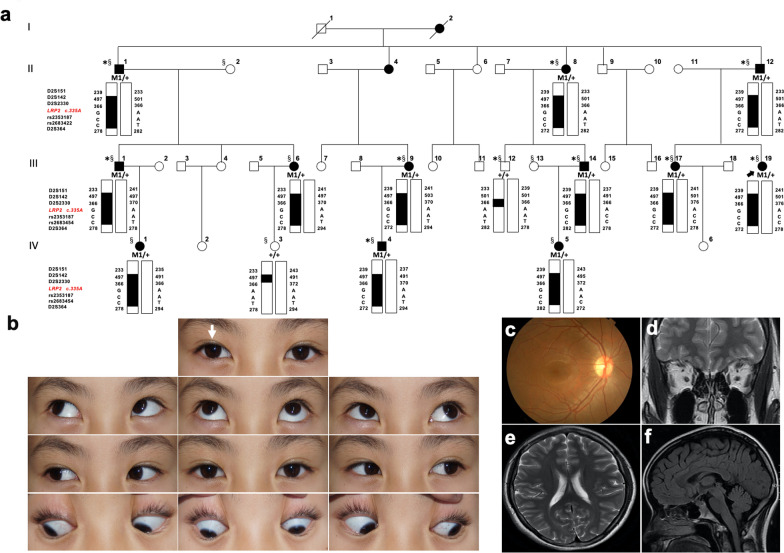
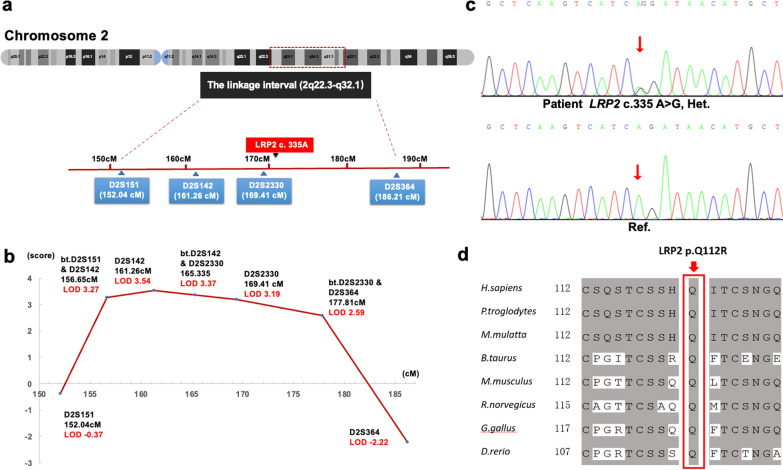
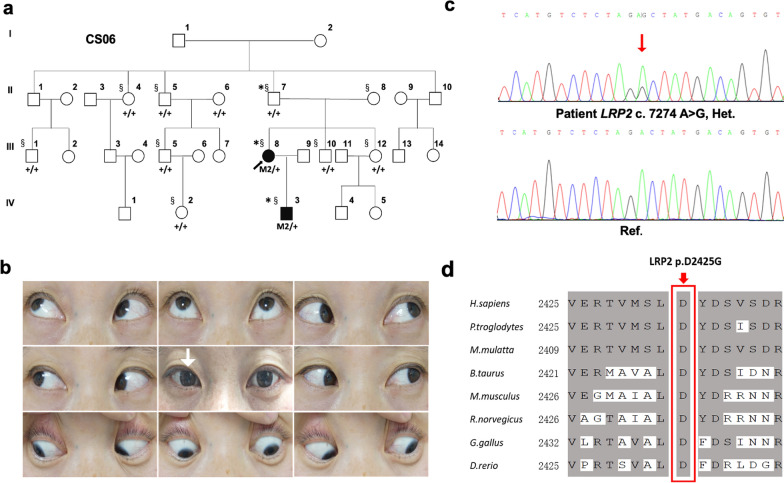
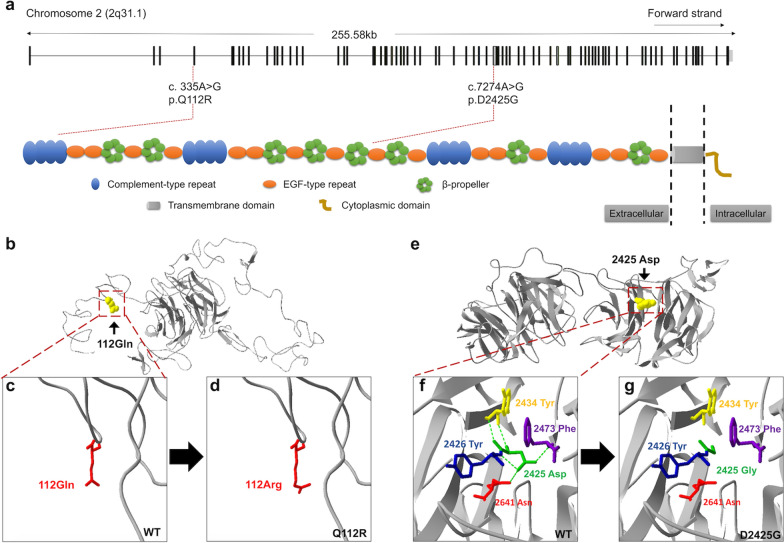
Results: Based on linkage analysis, CS in family CS08 mapped to a novel region of 34.17 centimorgan (cM) on chromosome 2q22.3-2q32.1 between markers D2S151 and D2S364, with a maximum log odds (LOD) score of 3.54 (theta = 0) at D2S142. Parallel WES identified a heterozygous variant, LRP2 c.335 A > G (p.Q112R), located in such a linkage interval that completely co-segregated with the disease in the family. Furthermore, another novel heterozygous variant (c.7274A > G, p.D2425G) in LRP2 that co-segregated was detected in 2 additional affected individuals from another unrelated family by WES. Both variants are predicted to be damaging by PolyPhen-2, SIFT and MutationTaster, and were absent in 100 ethnically matched normal controls.
Conclusion: LRP2 is a novel candidate genetic cause of non-syndromic familial CS.
Keywords: Comitant strabismus; Linkage analysis; Mutation; Phenotype; Whole-exome sequencing.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations.Hum Reprod. 2016 Dec;31(12):2872-2880. doi: 10.1093/humrep/dew262. Epub 2016 Oct 26. Hum Reprod. 2016. PMID: 27798045
-
Identification of a novel MYO6 mutation associated with autosomal dominant non-syndromic hearing loss in a Chinese family by whole-exome sequencing.Genes Genet Syst. 2018 Dec 22;93(5):171-179. doi: 10.1266/ggs.18-00006. Epub 2018 Aug 31. Genes Genet Syst. 2018. PMID: 30175721
-
Linkage and exome analysis implicate multiple genes in non-syndromic intellectual disability in a large Swedish family.BMC Med Genomics. 2019 Nov 6;12(1):156. doi: 10.1186/s12920-019-0606-4. BMC Med Genomics. 2019. PMID: 31694657 Free PMC article.
-
Identification of a homozygous missense mutation in LRP2 and a hemizygous missense mutation in TSPYL2 in a family with mild intellectual disability.Psychiatr Genet. 2016 Apr;26(2):66-73. doi: 10.1097/YPG.0000000000000114. Psychiatr Genet. 2016. PMID: 26529358
-
Role of Abelson Helper Integration Site 1, Nebulin, and Paired Box 3 Genes in the Development of Nonsyndromic Strabismus in a Series of Iranian Families: Sequence Analysis and Systematic Review of the Genetics of Nonsyndromic Strabismus.J Curr Ophthalmol. 2024 Mar 29;35(3):216-225. doi: 10.4103/joco.joco_53_22. eCollection 2023 Jul-Sep. J Curr Ophthalmol. 2024. PMID: 38681684 Free PMC article. Review.
Cited by
-
Genetics of strabismus.Front Ophthalmol (Lausanne). 2023;3:1233866. doi: 10.3389/fopht.2023.1233866. Epub 2023 Jul 20. Front Ophthalmol (Lausanne). 2023. PMID: 38500555 Free PMC article.
-
Developmental trends and knowledge frameworks in the application of radiomics in prostate cancer: a bibliometric analysis from 2000 to 2024.Discov Oncol. 2024 Dec 18;15(1):781. doi: 10.1007/s12672-024-01678-7. Discov Oncol. 2024. PMID: 39692833 Free PMC article.
-
Morphometric Analysis of the Eye by Magnetic Resonance Imaging in MGST2-Gene-Deficient Mice.Biomedicines. 2024 Feb 5;12(2):370. doi: 10.3390/biomedicines12020370. Biomedicines. 2024. PMID: 38397974 Free PMC article.
-
Exploring WNT2 polymorphisms in comitant strabismus: A genetic association study.Gene. 2024 Nov 30;928:148797. doi: 10.1016/j.gene.2024.148797. Epub 2024 Jul 26. Gene. 2024. PMID: 39068999
References
Publication types
MeSH terms
Substances
Grants and funding
- 81673198/Innovative Research Group Project of the National Natural Science Foundation of China
- BK20161595/Natural Science Foundation of Jiangsu Province
- QNRC2016563/Key Laboratory of Jiangsu Province for Chemical Pollution Control and Resources Reuse (CN)
- SJCX20_0477/Postgraduate Research & Practice Innovation Program of Jiangsu Province
LinkOut - more resources
Full Text Sources
