

The SEQC2 epigenomics quality control (EpiQC) study
- PMID: 34872606
- PMCID: PMC8650396
- DOI: 10.1186/s13059-021-02529-2
The SEQC2 epigenomics quality control (EpiQC) study
Erratum in
-
Author Correction: The SEQC2 epigenomics quality control (EpiQC) study.Genome Biol. 2021 Dec 23;22(1):350. doi: 10.1186/s13059-021-02573-y. Genome Biol. 2021. PMID: 34949218 Free PMC article. No abstract available.
-
Author Correction: Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline.Genome Biol. 2022 Mar 8;23(1):76. doi: 10.1186/s13059-022-02645-7. Genome Biol. 2022. PMID: 35260190 Free PMC article. No abstract available.
Abstract
Background: Cytosine modifications in DNA such as 5-methylcytosine (5mC) underlie a broad range of developmental processes, maintain cellular lineage specification, and can define or stratify types of cancer and other diseases. However, the wide variety of approaches available to interrogate these modifications has created a need for harmonized materials, methods, and rigorous benchmarking to improve genome-wide methylome sequencing applications in clinical and basic research. Here, we present a multi-platform assessment and cross-validated resource for epigenetics research from the FDA's Epigenomics Quality Control Group.
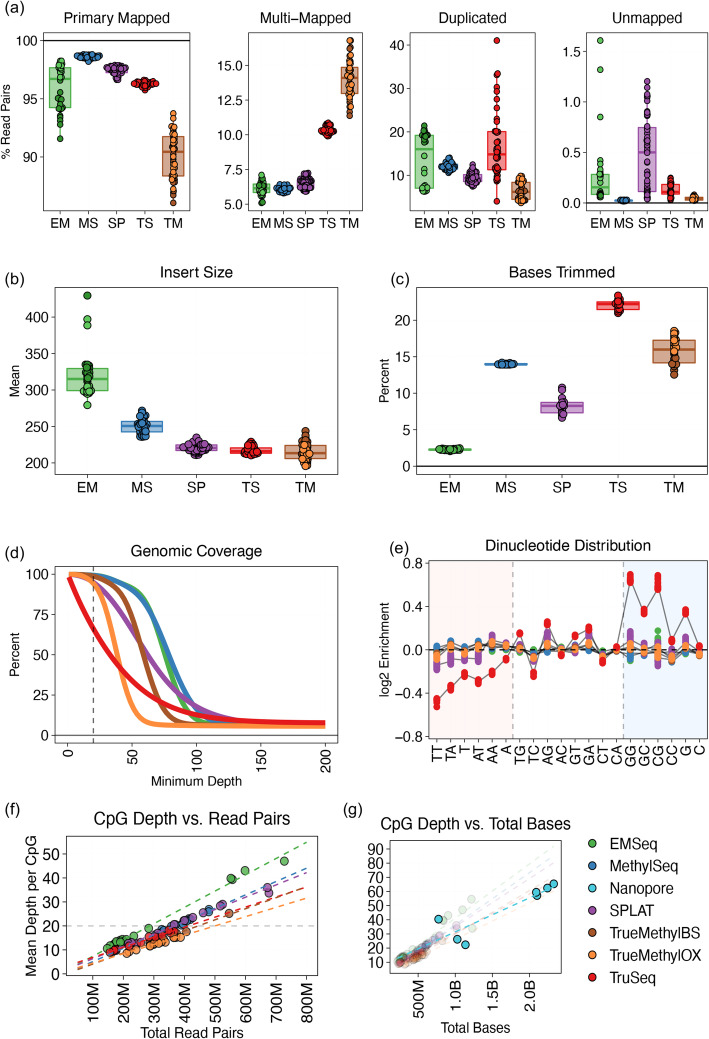
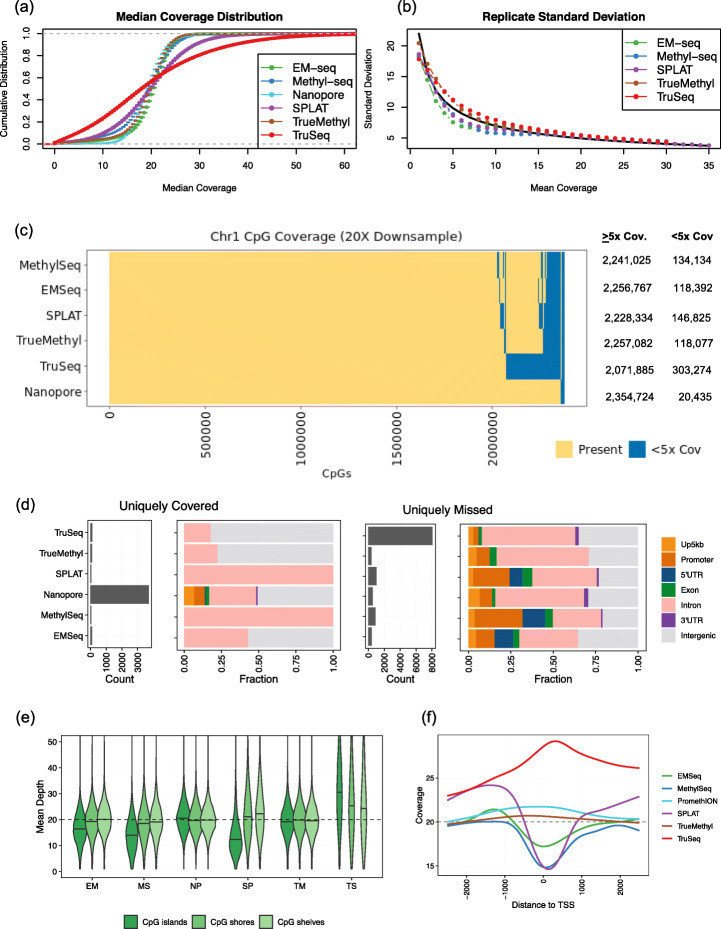
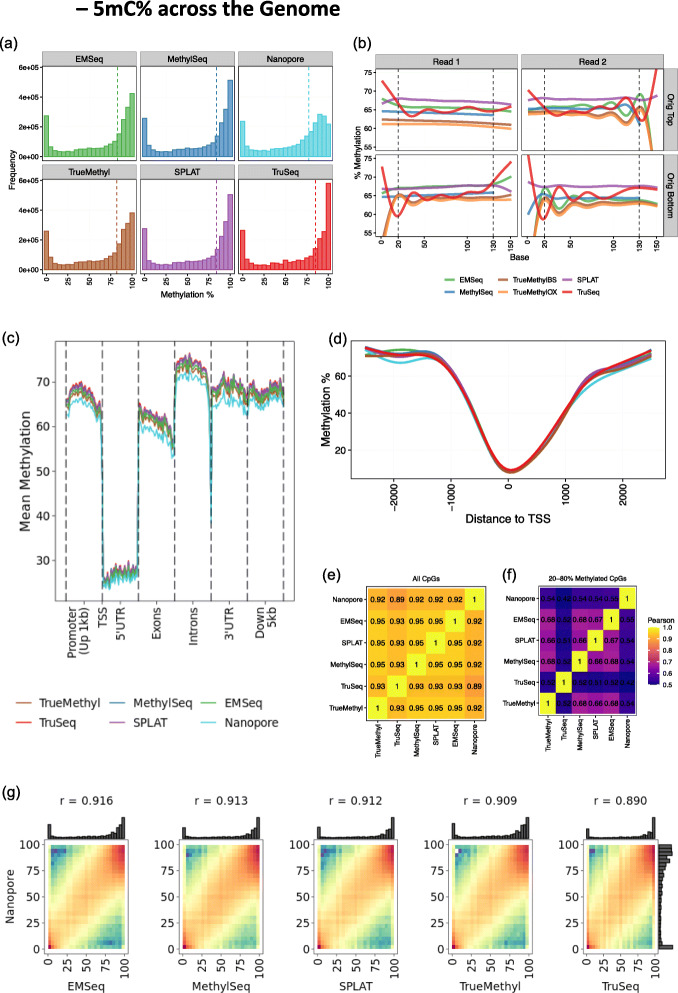
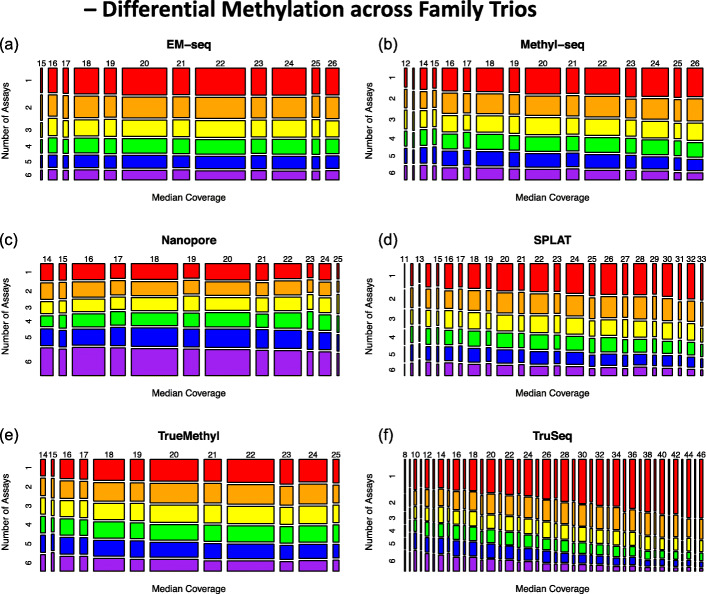
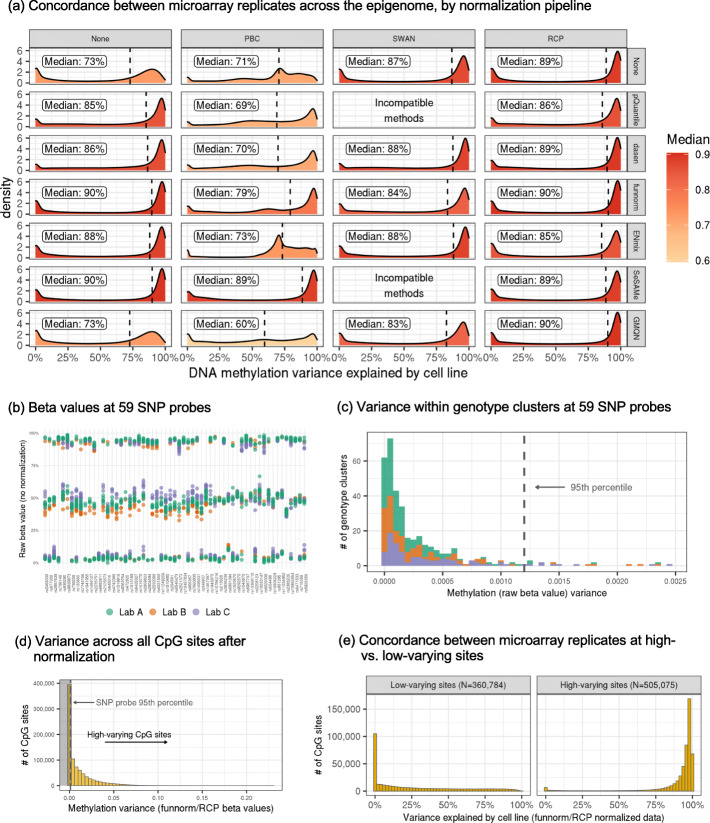
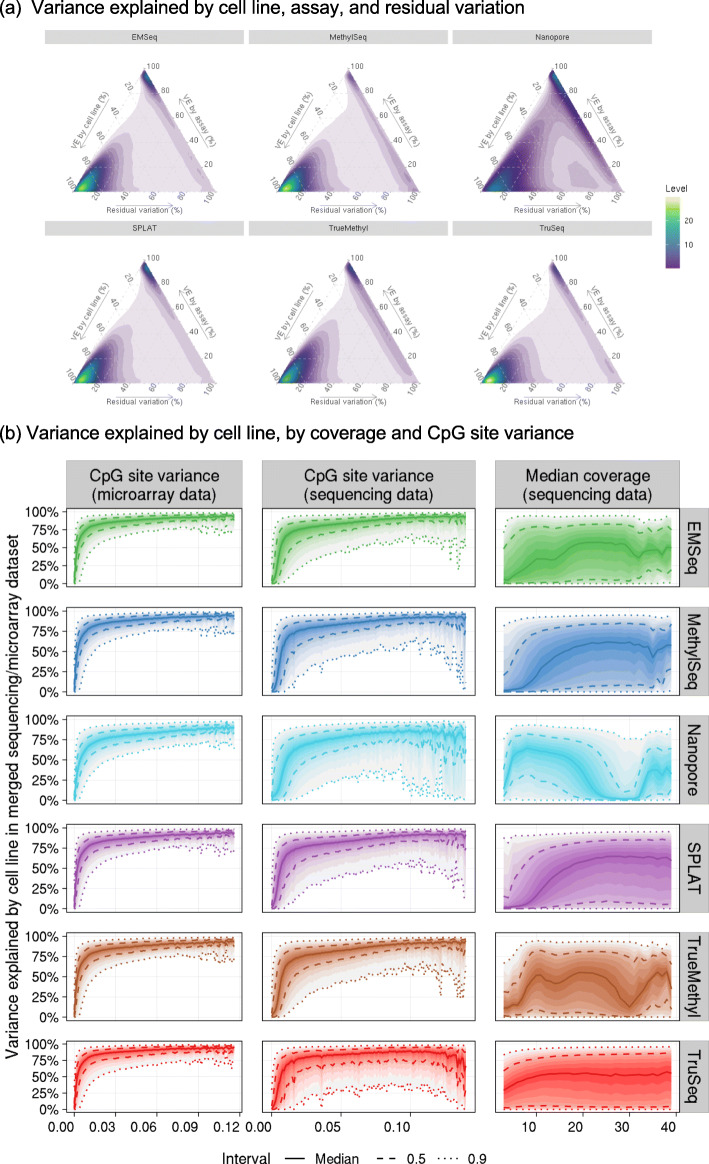
Results: Each sample is processed in multiple replicates by three whole-genome bisulfite sequencing (WGBS) protocols (TruSeq DNA methylation, Accel-NGS MethylSeq, and SPLAT), oxidative bisulfite sequencing (TrueMethyl), enzymatic deamination method (EMSeq), targeted methylation sequencing (Illumina Methyl Capture EPIC), single-molecule long-read nanopore sequencing from Oxford Nanopore Technologies, and 850k Illumina methylation arrays. After rigorous quality assessment and comparison to Illumina EPIC methylation microarrays and testing on a range of algorithms (Bismark, BitmapperBS, bwa-meth, and BitMapperBS), we find overall high concordance between assays, but also differences in efficiency of read mapping, CpG capture, coverage, and platform performance, and variable performance across 26 microarray normalization algorithms.
Conclusions: The data provided herein can guide the use of these DNA reference materials in epigenomics research, as well as provide best practices for experimental design in future studies. By leveraging seven human cell lines that are designated as publicly available reference materials, these data can be used as a baseline to advance epigenomics research.
© 2021. The Author(s).
Conflict of interest statement
B.W.L, M.C., L.W., and V.K.C.P are employees of New England Biolabs. S.L and J.W.D are employees of Abbvie, Inc. S.B is an employee of Illumina, Inc. F.W, J.I, and W.L are employees of New York Genome Center.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA249054/CA/NCI NIH HHS/United States
- R01 CA230514/CA/NCI NIH HHS/United States
- P50 HD105352/HD/NICHD NIH HHS/United States
- P20 GM103466/GM/NIGMS NIH HHS/United States
- U54 MD007601/MD/NIMHD NIH HHS/United States
- P30 GM114737/GM/NIGMS NIH HHS/United States
- R01 AI151059/AI/NIAID NIH HHS/United States
- P01 CA214274/CA/NCI NIH HHS/United States
- R01 CA223490/CA/NCI NIH HHS/United States
- P30 CA071789/CA/NCI NIH HHS/United States
- U01 DA053941/DA/NIDA NIH HHS/United States
- R01 MH117406/MH/NIMH NIH HHS/United States
- S10 OD019960/OD/NIH HHS/United States
- U54 MD007584/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
