

Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction
- PMID: 34873061
- PMCID: PMC8670487
- DOI: 10.1073/pnas.2110828118
Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction
Abstract
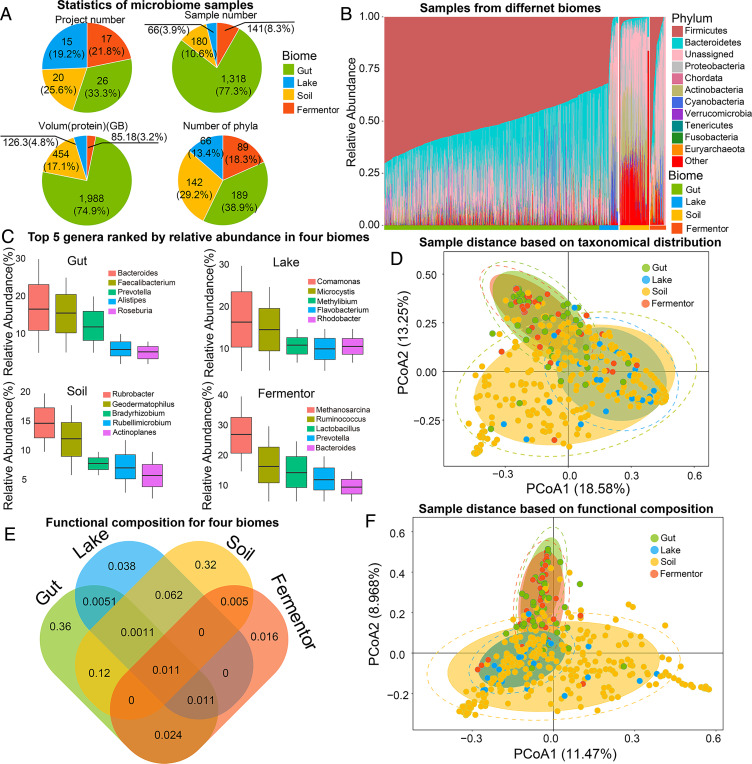
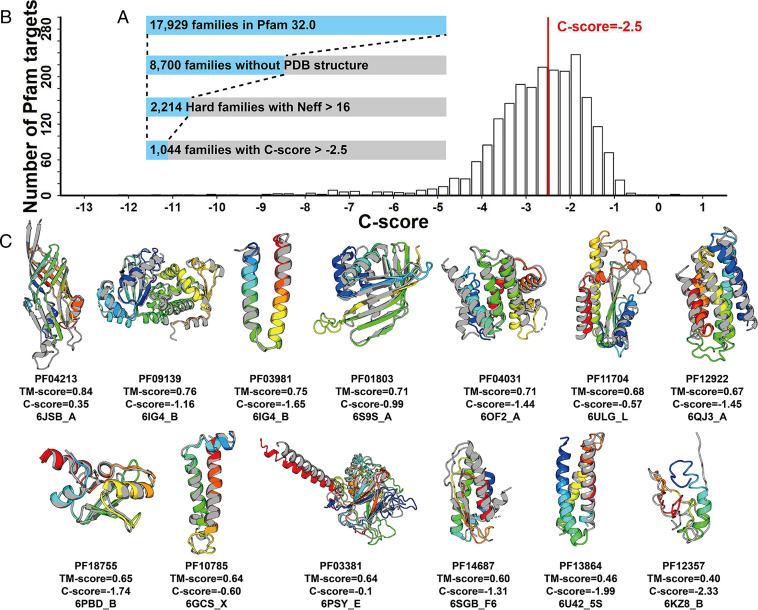
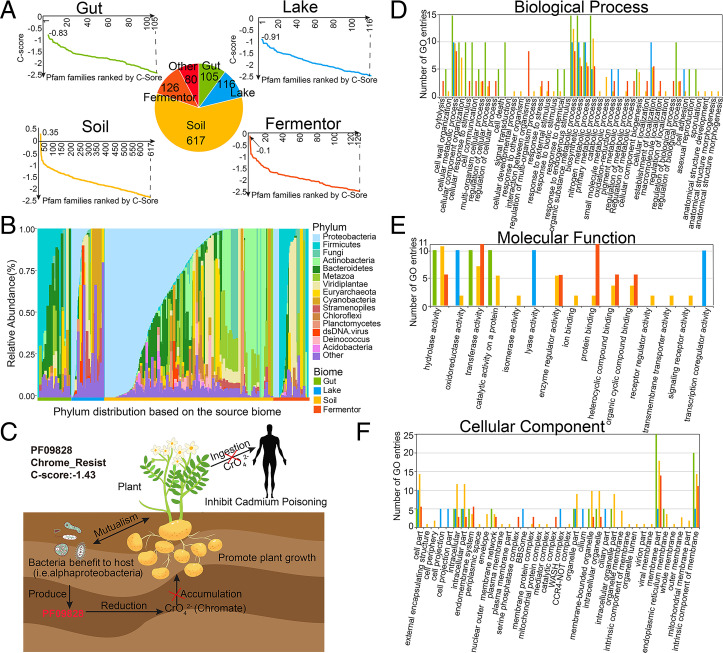
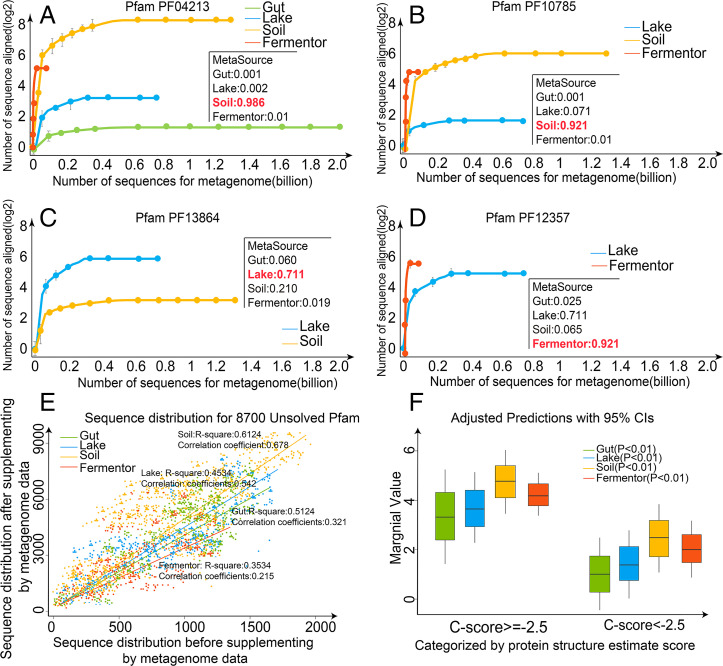
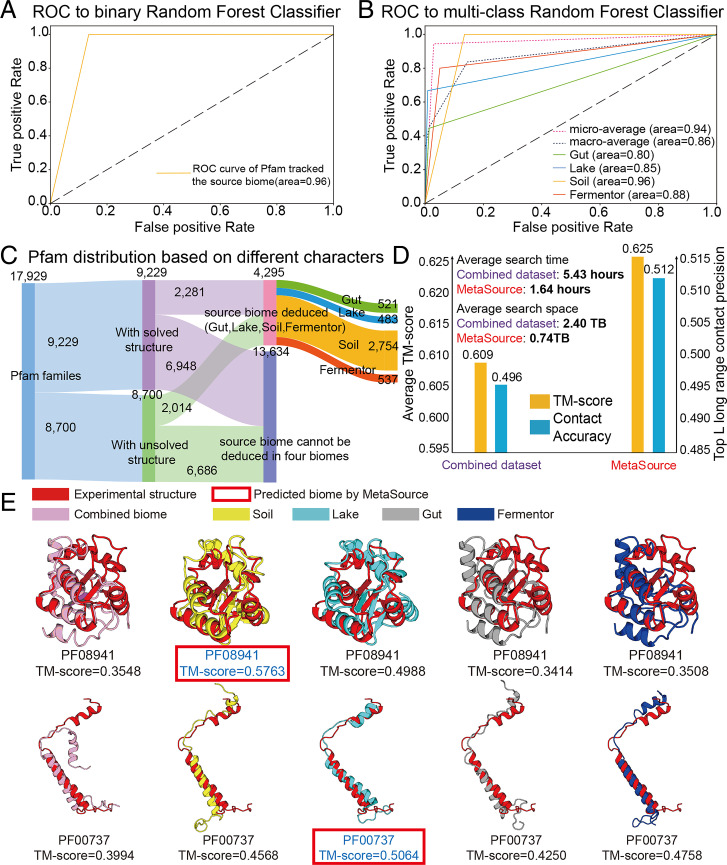
Information derived from metagenome sequences through deep-learning techniques has significantly improved the accuracy of template free protein structure modeling. However, most of the deep learning-based modeling studies are based on blind sequence database searches and suffer from low efficiency in computational resource utilization and model construction, especially when the sequence library becomes prohibitively large. We proposed a MetaSource model built on 4.25 billion microbiome sequences from four major biomes (Gut, Lake, Soil, and Fermentor) to decode the inherent linkage of microbial niches with protein homologous families. Large-scale protein family folding experiments on 8,700 unknown Pfam families showed that a microbiome targeted approach with multiple sequence alignment constructed from individual MetaSource biomes requires more than threefold less computer memory and CPU (central processing unit) time but generates contact-map and three-dimensional structure models with a significantly higher accuracy, compared with that using combined metagenome datasets. These results demonstrate an avenue to bridge the gap between the rapidly increasing metagenome databases and the limited computing resources for efficient genome-wide database mining, which provides a useful bluebook to guide future microbiome sequence database and modeling development for high-accuracy protein structure and function prediction.
Keywords: deep learning; microbiome; multiple sequence alignments; protein homologous families; protein structure prediction.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13.Proteins. 2019 Dec;87(12):1082-1091. doi: 10.1002/prot.25798. Epub 2019 Aug 22. Proteins. 2019. PMID: 31407406 Free PMC article.
-
Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13.Proteins. 2019 Dec;87(12):1165-1178. doi: 10.1002/prot.25697. Epub 2019 Apr 25. Proteins. 2019. PMID: 30985027 Free PMC article.
-
Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model.PLoS Comput Biol. 2017 Jan 5;13(1):e1005324. doi: 10.1371/journal.pcbi.1005324. eCollection 2017 Jan. PLoS Comput Biol. 2017. PMID: 28056090 Free PMC article.
-
A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction.Curr Opin Struct Biol. 2005 Jun;15(3):285-9. doi: 10.1016/j.sbi.2005.05.011. Curr Opin Struct Biol. 2005. PMID: 15939584 Review.
-
Deep Learning-Based Advances in Protein Structure Prediction.Int J Mol Sci. 2021 May 24;22(11):5553. doi: 10.3390/ijms22115553. Int J Mol Sci. 2021. PMID: 34074028 Free PMC article. Review.
Cited by
-
Petascale Homology Search for Structure Prediction.bioRxiv [Preprint]. 2023 Jul 11:2023.07.10.548308. doi: 10.1101/2023.07.10.548308. bioRxiv. 2023. Update in: Cold Spring Harb Perspect Biol. 2024 May 2;16(5):a041465. doi: 10.1101/cshperspect.a041465. PMID: 37503235 Free PMC article. Updated. Preprint.
-
Characterization of Treponema denticola Major Surface Protein (Msp) by Deletion Analysis and Advanced Molecular Modeling.J Bacteriol. 2022 Sep 20;204(9):e0022822. doi: 10.1128/jb.00228-22. Epub 2022 Aug 1. J Bacteriol. 2022. PMID: 35913147 Free PMC article.
-
Improving AlphaFold2- and AlphaFold3-Based Protein Complex Structure Prediction With MULTICOM4 in CASP16.Proteins. 2025 Jun 2:10.1002/prot.26850. doi: 10.1002/prot.26850. Online ahead of print. Proteins. 2025. PMID: 40452318
-
Designing of thiazolidinones against chicken pox, monkey pox, and hepatitis viruses: A computational approach.Comput Biol Chem. 2023 Apr;103:107827. doi: 10.1016/j.compbiolchem.2023.107827. Epub 2023 Feb 12. Comput Biol Chem. 2023. PMID: 36805155 Free PMC article.
-
Integrating deep learning, threading alignments, and a multi-MSA strategy for high-quality protein monomer and complex structure prediction in CASP15.Proteins. 2023 Dec;91(12):1684-1703. doi: 10.1002/prot.26585. Epub 2023 Aug 31. Proteins. 2023. PMID: 37650367 Free PMC article.
References
-
- Baker D., Sali A., Protein structure prediction and structural genomics. Science 294, 93–96 (2001). - PubMed
-
- Simons K. T., Kooperberg C., Huang E., Baker D., Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
