

Proximal and distal effects of genetic susceptibility to multiple sclerosis on the T cell epigenome
- PMID: 34873174
- PMCID: PMC8648735
- DOI: 10.1038/s41467-021-27427-w
Proximal and distal effects of genetic susceptibility to multiple sclerosis on the T cell epigenome
Abstract
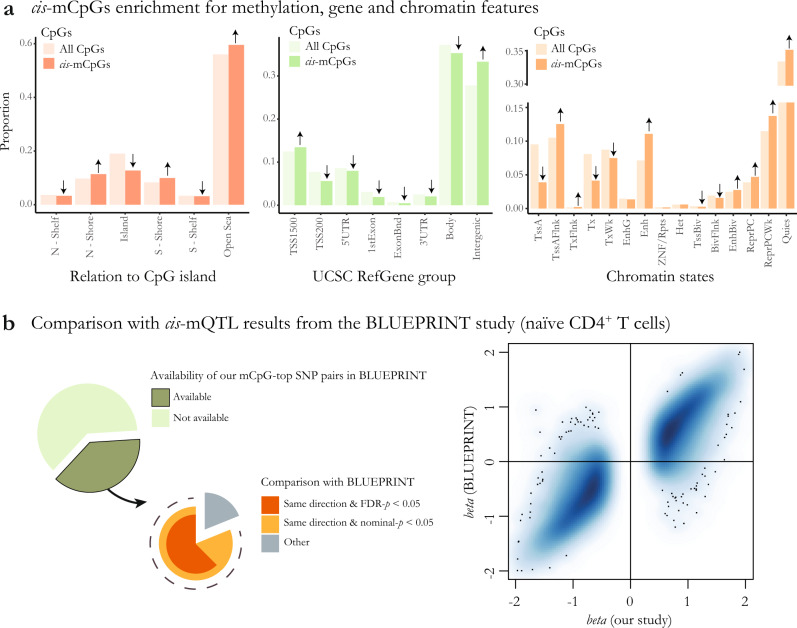
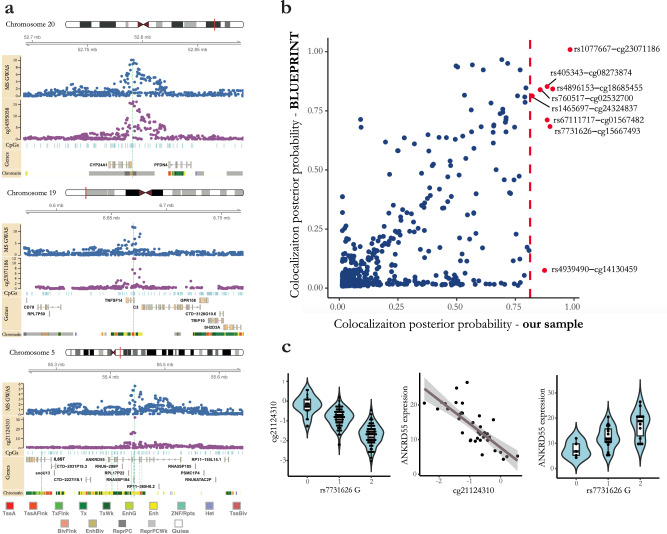
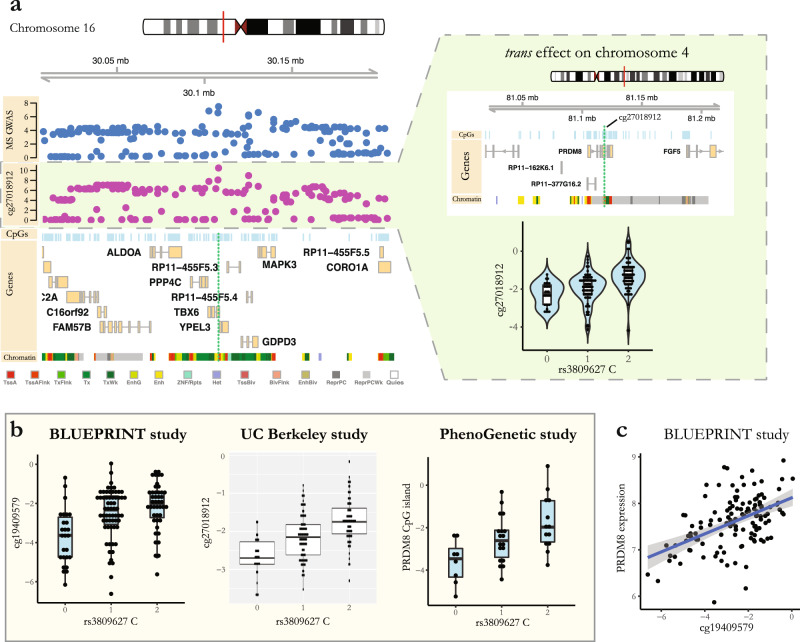
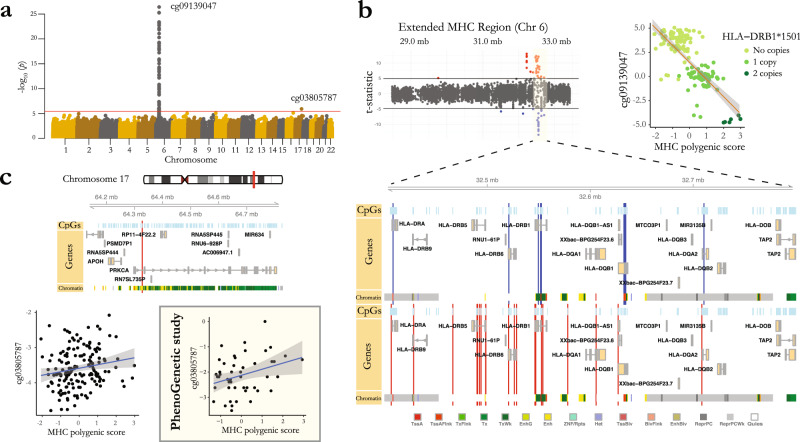
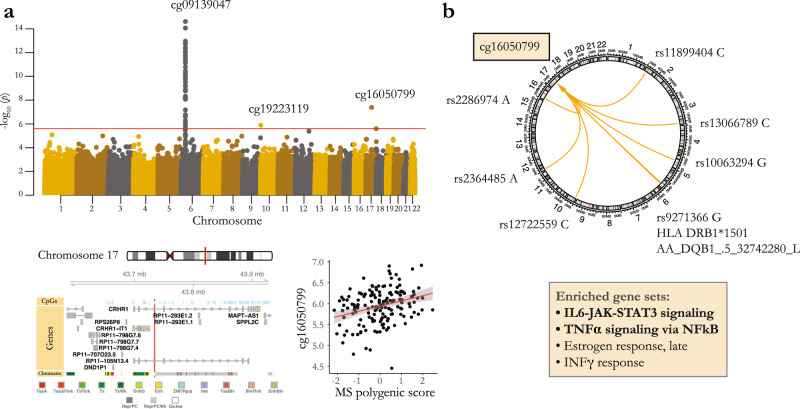
Identifying the effects of genetic variation on the epigenome in disease-relevant cell types can help advance our understanding of the first molecular contributions of genetic susceptibility to disease onset. Here, we establish a genome-wide map of DNA methylation quantitative trait loci in CD4+ T-cells isolated from multiple sclerosis patients. Utilizing this map in a colocalization analysis, we identify 19 loci where the same haplotype drives both multiple sclerosis susceptibility and local DNA methylation. We also identify two distant methylation effects of multiple sclerosis susceptibility loci: a chromosome 16 locus affects PRDM8 methylation (a chromosome 4 region not previously associated with multiple sclerosis), and the aggregate effect of multiple sclerosis-associated variants in the major histocompatibility complex influences DNA methylation near PRKCA (chromosome 17). Overall, we present a new resource for a key cell type in inflammatory disease research and uncover new gene targets for the study of predisposition to multiple sclerosis.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis.Nature. 2010 Apr 29;464(7293):1351-6. doi: 10.1038/nature08990. Nature. 2010. PMID: 20428171 Free PMC article.
-
Multiple Sclerosis Risk Allele in CLEC16A Acts as an Expression Quantitative Trait Locus for CLEC16A and SOCS1 in CD4+ T Cells.PLoS One. 2015 Jul 23;10(7):e0132957. doi: 10.1371/journal.pone.0132957. eCollection 2015. PLoS One. 2015. PMID: 26203907 Free PMC article.
-
Increased THEMIS First Exon Usage in CD4+ T-Cells Is Associated with a Genotype that Is Protective against Multiple Sclerosis.PLoS One. 2016 Jul 20;11(7):e0158327. doi: 10.1371/journal.pone.0158327. eCollection 2016. PLoS One. 2016. PMID: 27438997 Free PMC article.
-
Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era.Genome Biol. 2017 Jun 19;18(1):120. doi: 10.1186/s13059-017-1250-y. Genome Biol. 2017. PMID: 28629478 Free PMC article. Review.
-
The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years.Trends Genet. 2017 Dec;33(12):960-970. doi: 10.1016/j.tig.2017.09.004. Epub 2017 Oct 5. Trends Genet. 2017. PMID: 28987266 Free PMC article. Review.
Cited by
-
Epigenetic control of ataxin-1 in multiple sclerosis.Ann Clin Transl Neurol. 2022 Aug;9(8):1186-1194. doi: 10.1002/acn3.51618. Epub 2022 Jul 28. Ann Clin Transl Neurol. 2022. PMID: 35903875 Free PMC article.
-
Association of ANKRD55 Gene Polymorphism with HT: A Protective Factor for Disease Susceptibility.Int J Endocrinol. 2022 Aug 9;2022:7300796. doi: 10.1155/2022/7300796. eCollection 2022. Int J Endocrinol. 2022. PMID: 35983018 Free PMC article.
-
Differential DNA methylation associated with multiple sclerosis and disease modifying treatments in an underrepresented minority population.Front Genet. 2023 Jan 4;13:1058817. doi: 10.3389/fgene.2022.1058817. eCollection 2022. Front Genet. 2023. PMID: 36685876 Free PMC article.
-
A scoping review of statistical methods to investigate colocalization between genetic associations and microRNA expression in osteoarthritis.Osteoarthr Cartil Open. 2024 Nov 8;6(4):100540. doi: 10.1016/j.ocarto.2024.100540. eCollection 2024 Dec. Osteoarthr Cartil Open. 2024. PMID: 39640910 Free PMC article.
-
Differential impact of environmental factors on systemic and localized autoimmunity.Front Immunol. 2023 May 22;14:1147447. doi: 10.3389/fimmu.2023.1147447. eCollection 2023. Front Immunol. 2023. PMID: 37283765 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
