

CHARMM-GUI Drude prepper for molecular dynamics simulation using the classical Drude polarizable force field
- PMID: 34874077
- PMCID: PMC8741736
- DOI: 10.1002/jcc.26795
CHARMM-GUI Drude prepper for molecular dynamics simulation using the classical Drude polarizable force field
Abstract
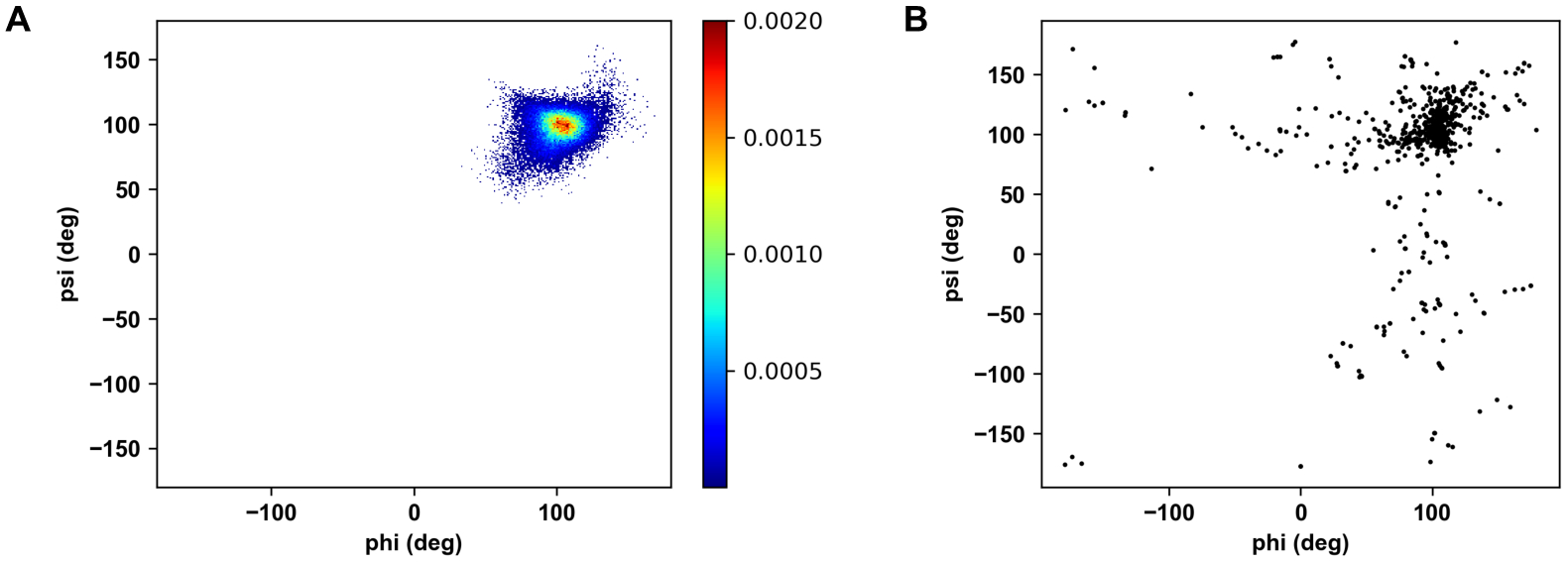
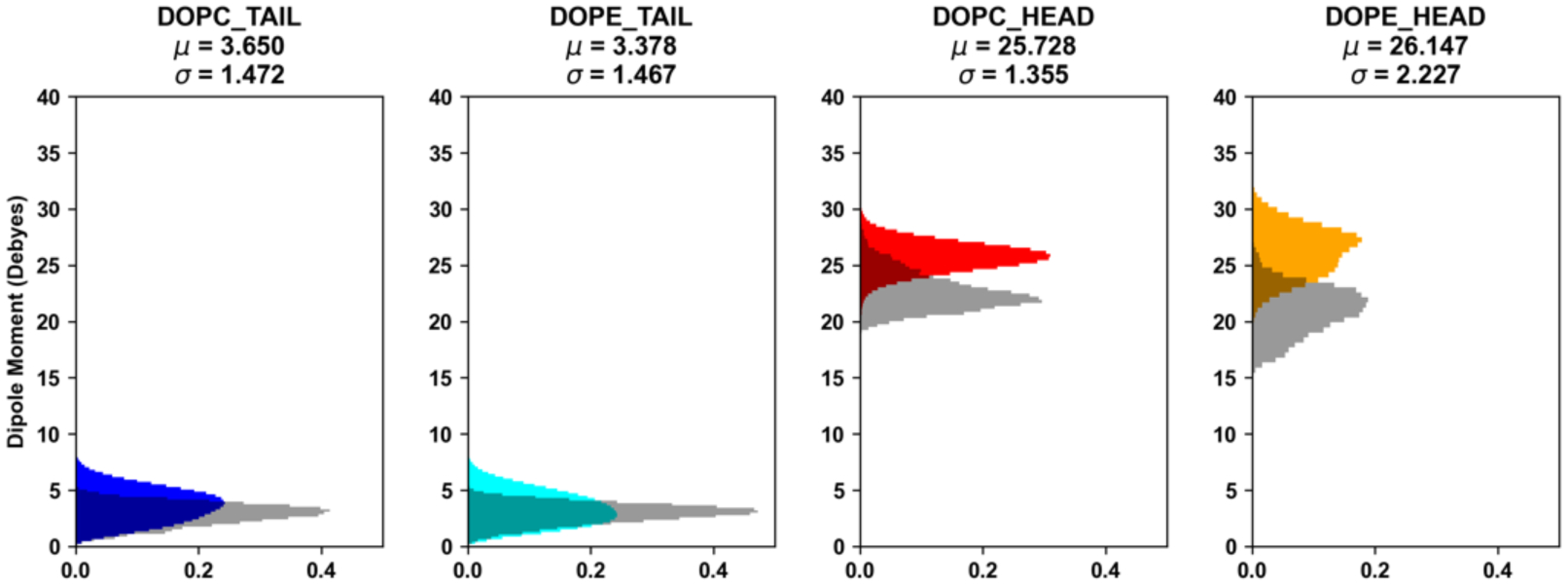
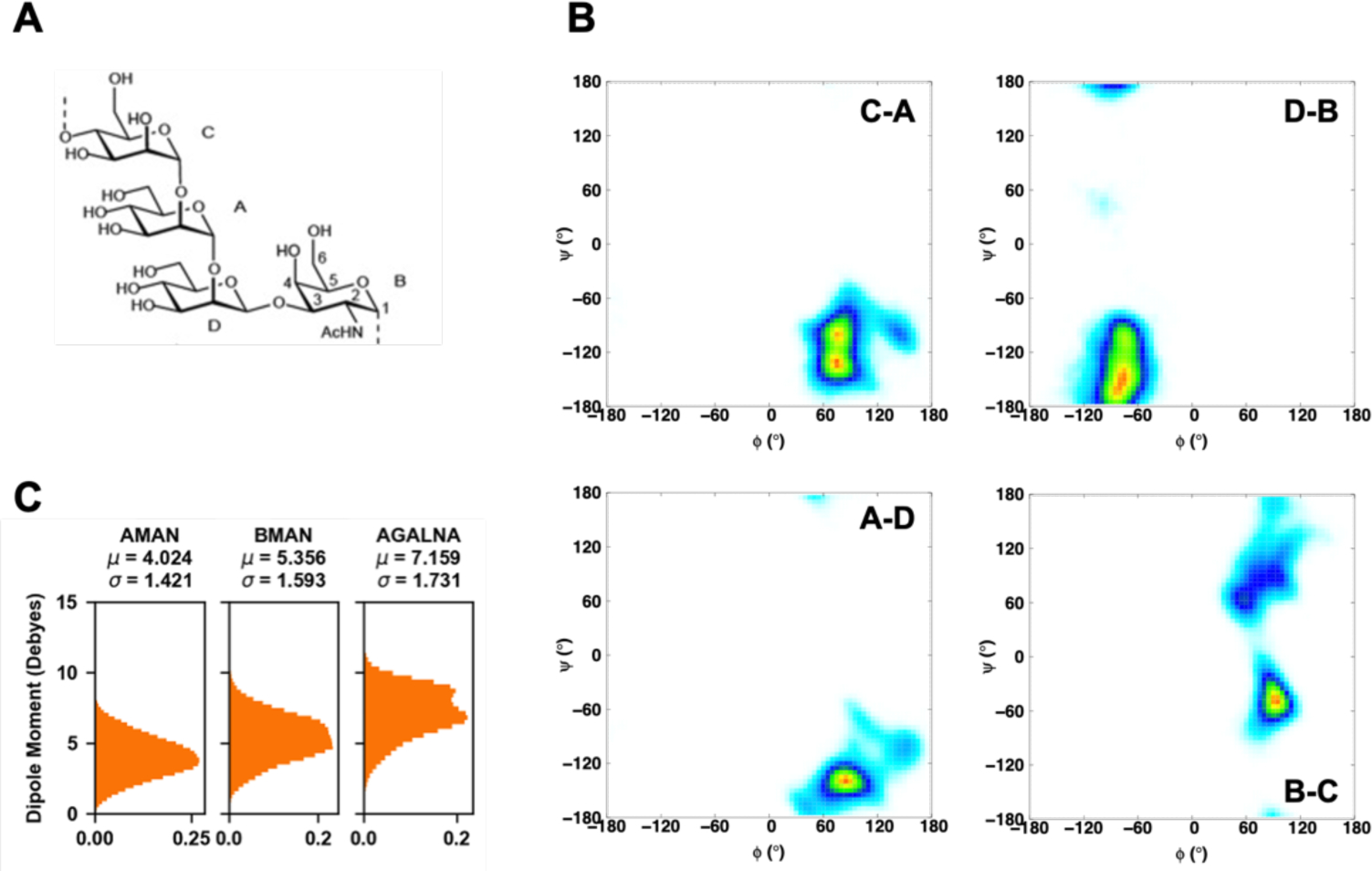
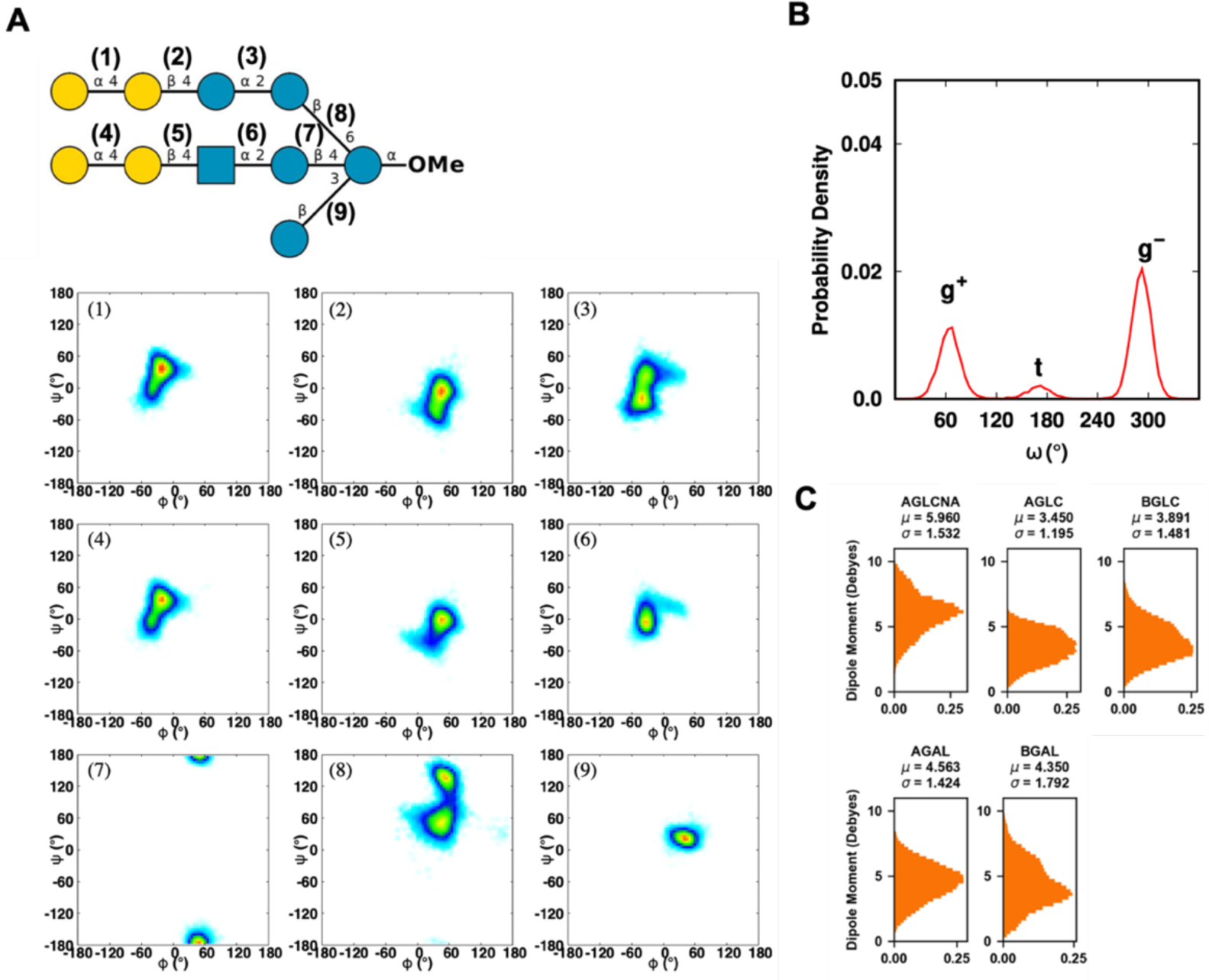
Explicit treatment of electronic polarizability in empirical force fields (FFs) represents an extension over a traditional additive or pairwise FF and provides a more realistic model of the variations in electronic structure in condensed phase, macromolecular simulations. To facilitate utilization of the polarizable FF based on the classical Drude oscillator model, Drude Prepper has been developed in CHARMM-GUI. Drude Prepper ingests additive CHARMM protein structures file (PSF) and pre-equilibrated coordinates in CHARMM, PDB, or NAMD format, from which the molecular components of the system are identified. These include all residues and patches connecting those residues along with water, ions, and other solute molecules. This information is then used to construct the Drude FF-based PSF using molecular generation capabilities in CHARMM, followed by minimization and equilibration. In addition, inputs are generated for molecular dynamics (MD) simulations using CHARMM, GROMACS, NAMD, and OpenMM. Validation of the Drude Prepper protocol and inputs is performed through conversion and MD simulations of various heterogeneous systems that include proteins, nucleic acids, lipids, polysaccharides, and atomic ions using the aforementioned simulation packages. Stable simulations are obtained in all studied systems, including 5 μs simulation of ubiquitin, verifying the integrity of the generated Drude PSFs. In addition, the ability of the Drude FF to model variations in electronic structure is shown through dipole moment analysis in selected systems. The capabilities and availability of Drude Prepper in CHARMM-GUI is anticipated to greatly facilitate the application of the Drude FF to a range of condensed phase, macromolecular systems.
Keywords: DNA; RNA; carbohydrate; dipole moment; membrane; protein; protein structure file.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
Figures
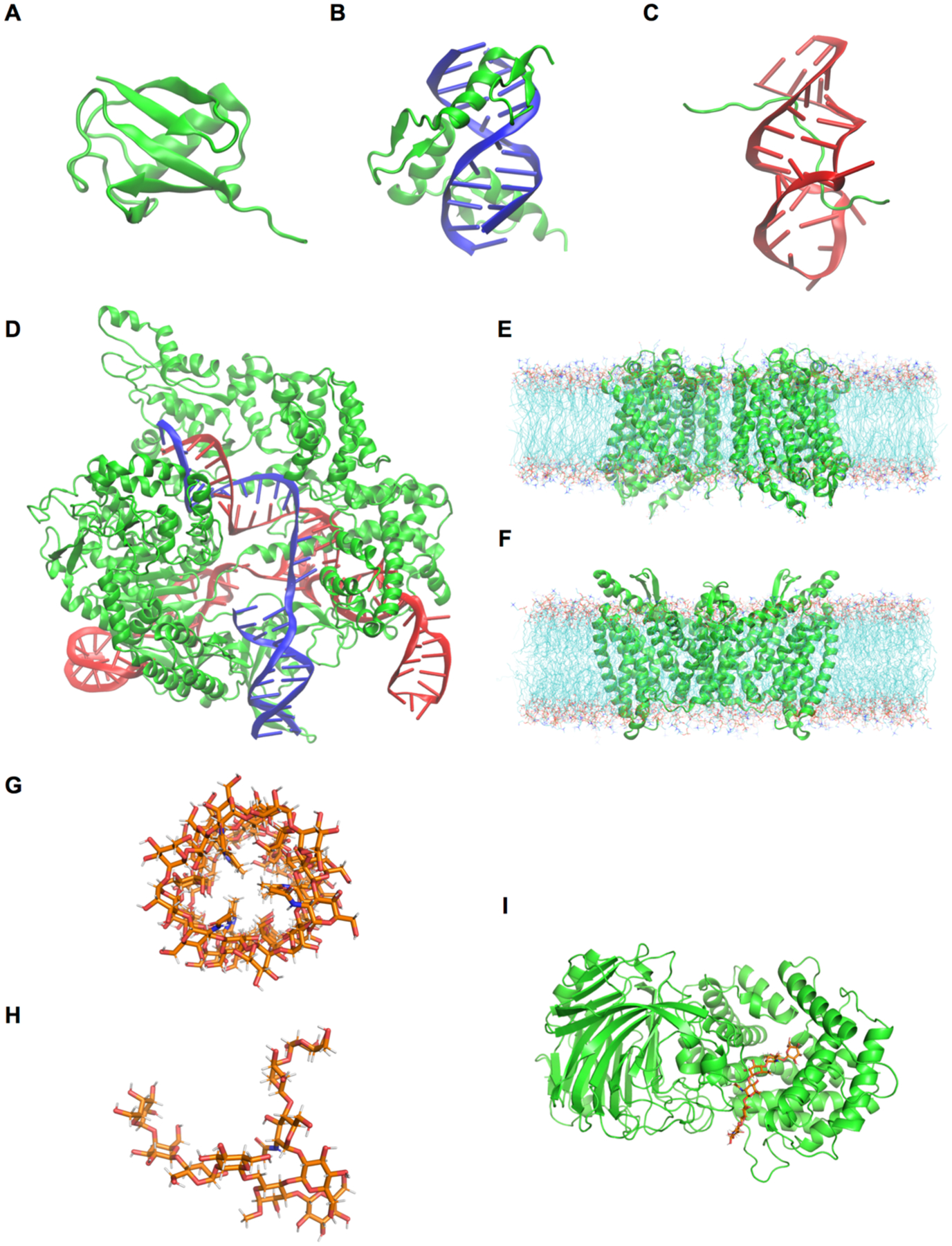
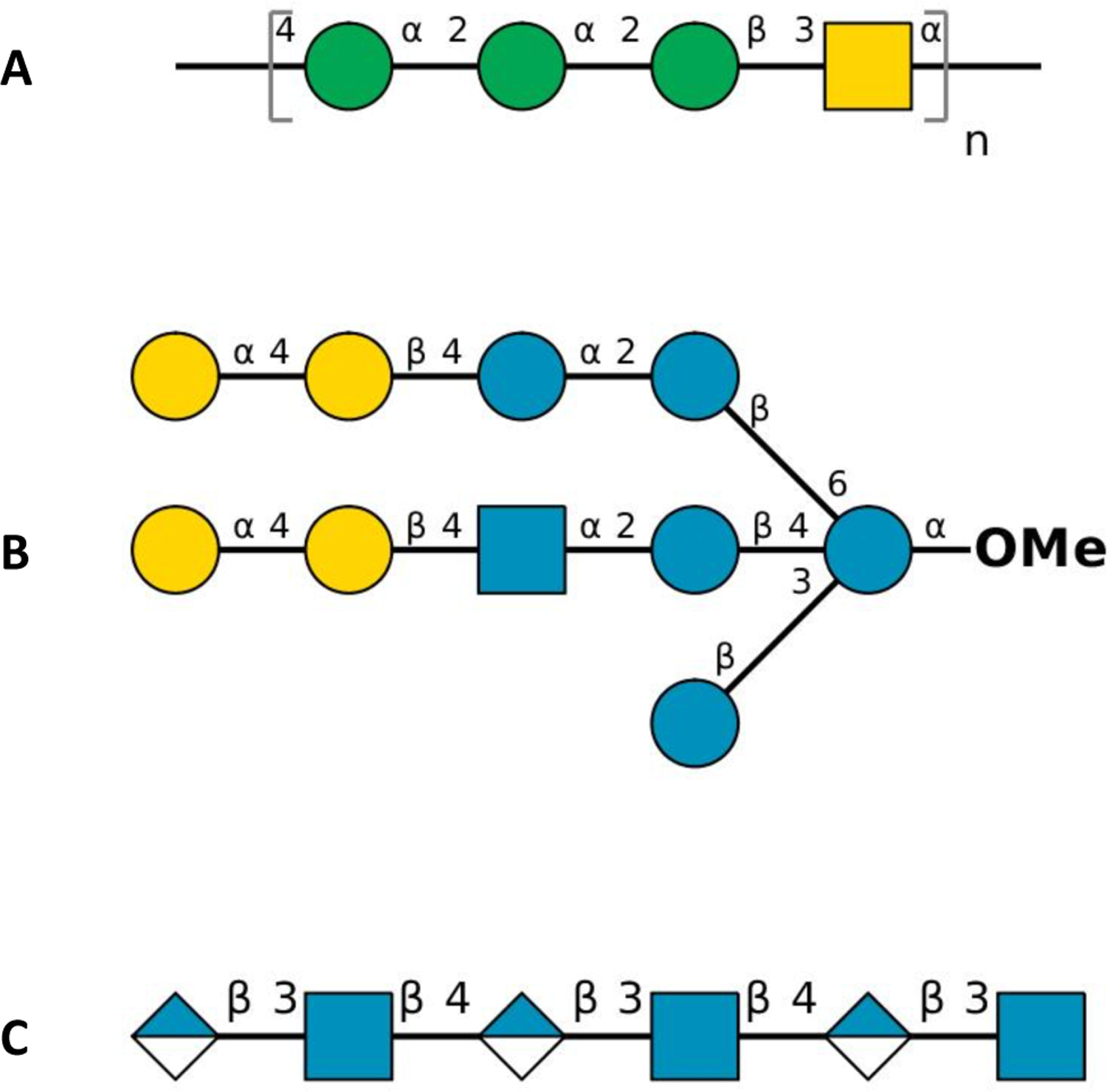
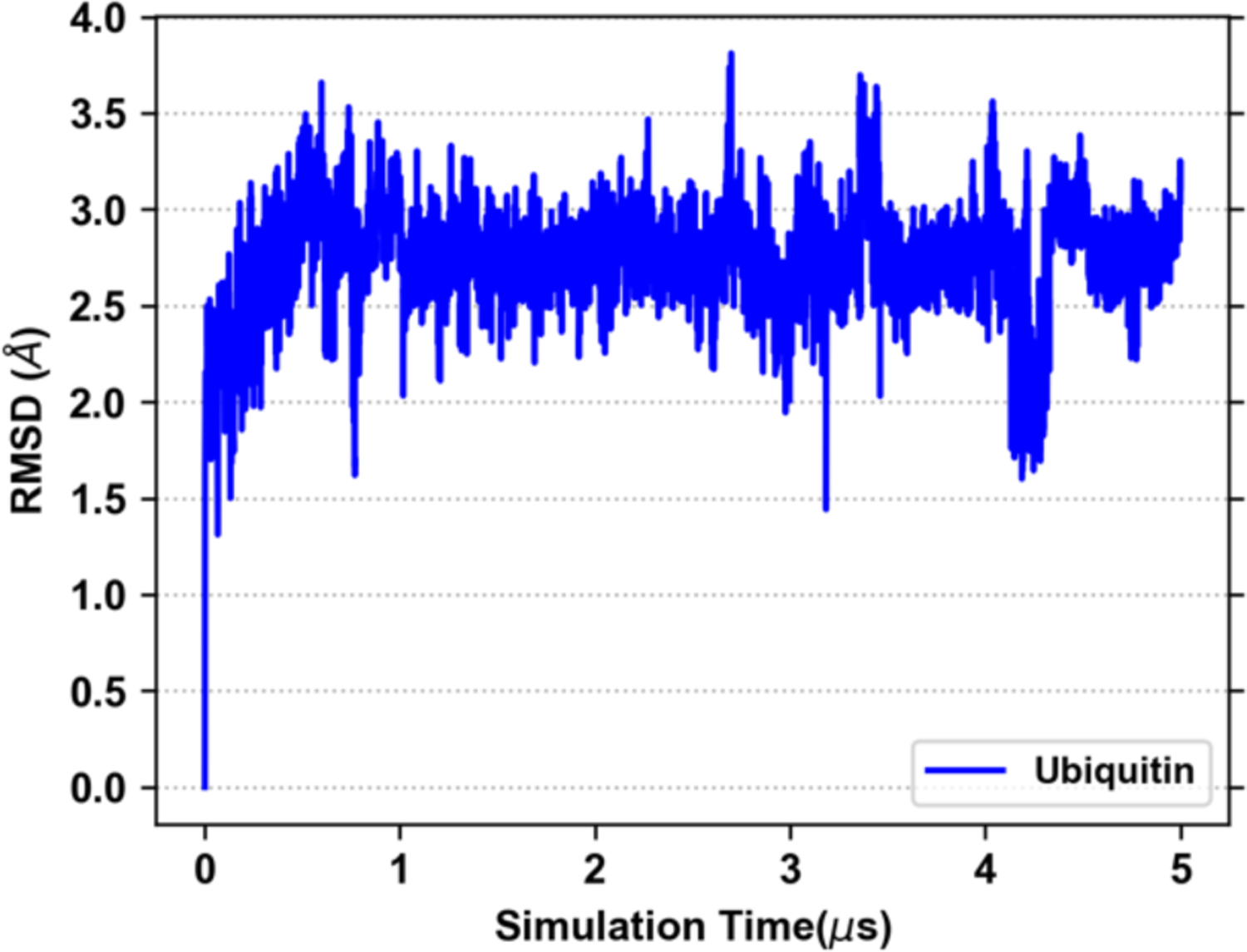
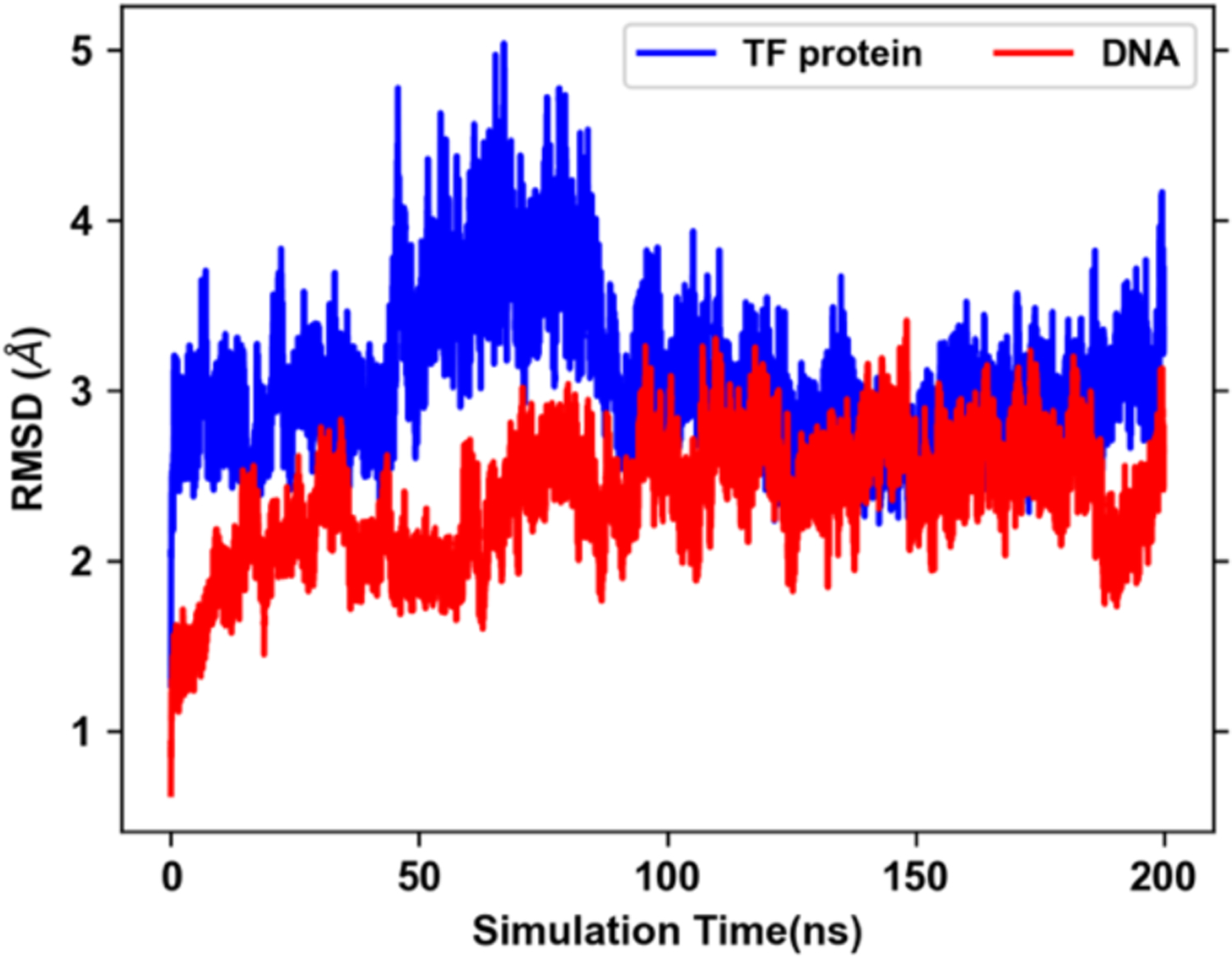
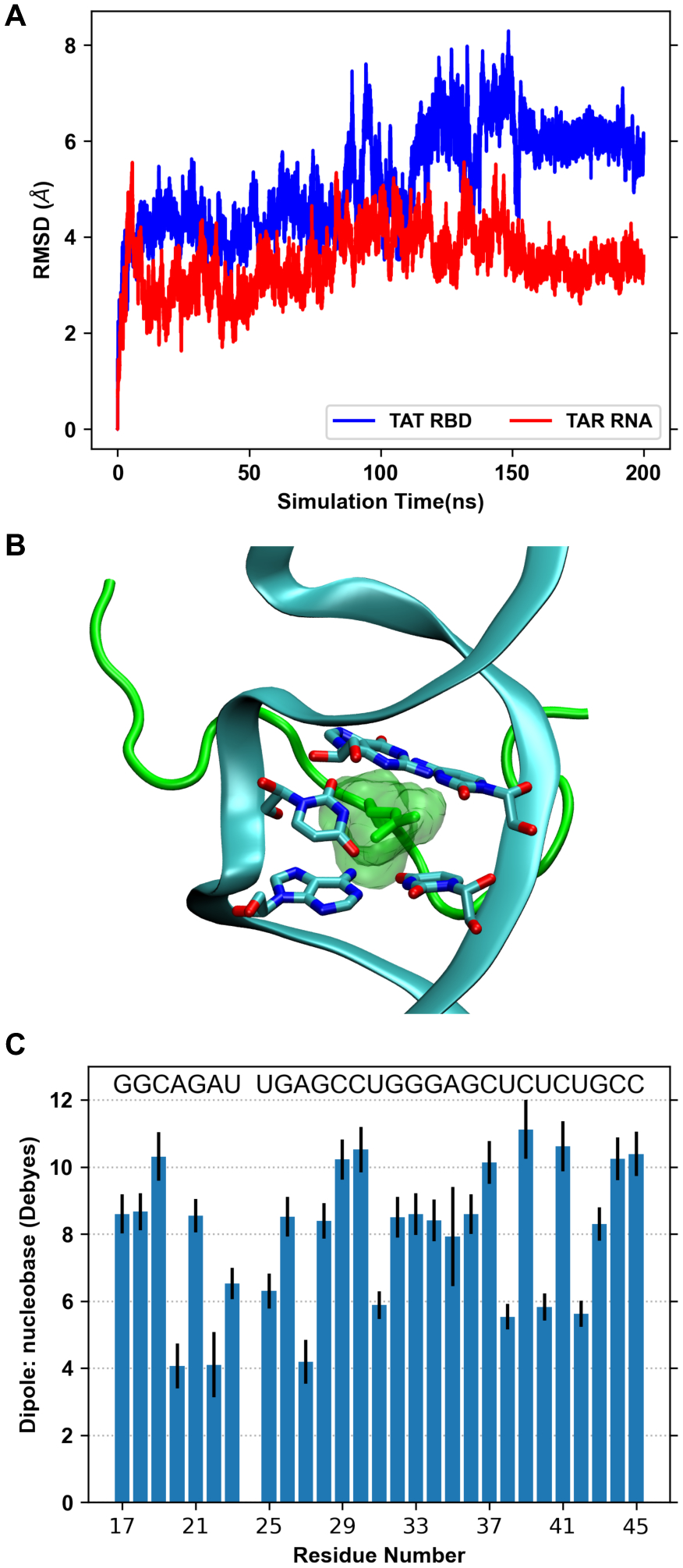
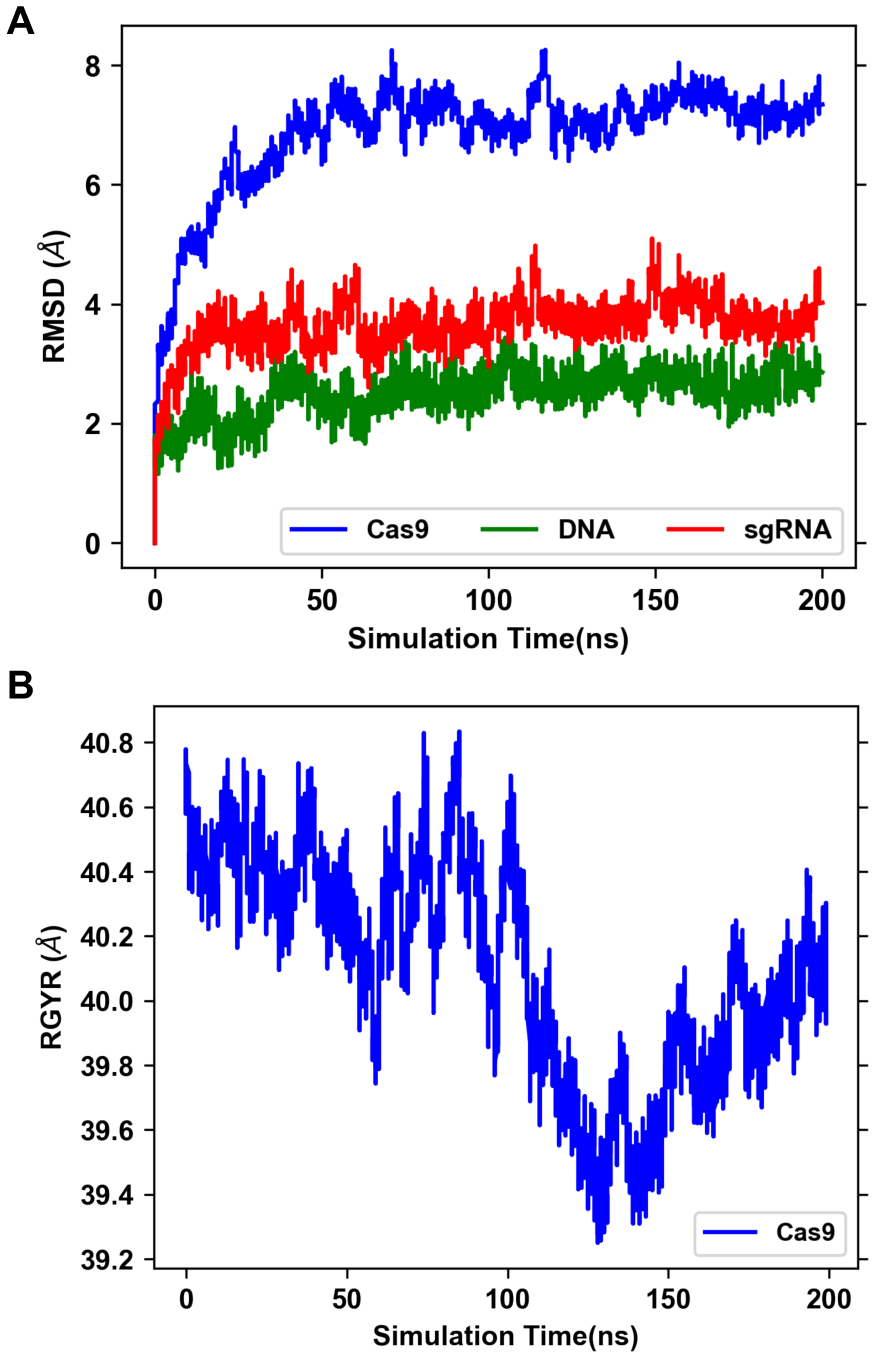
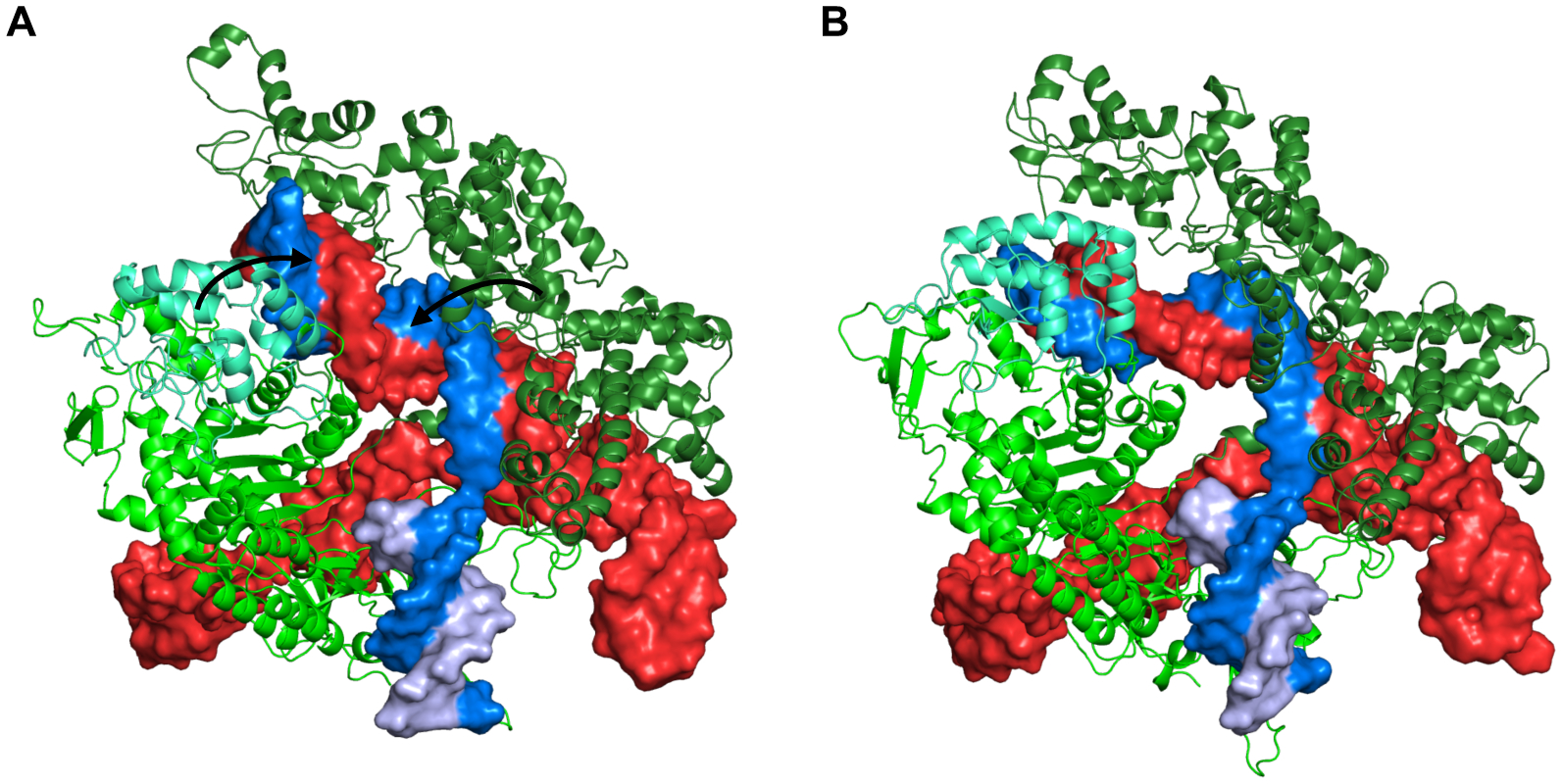
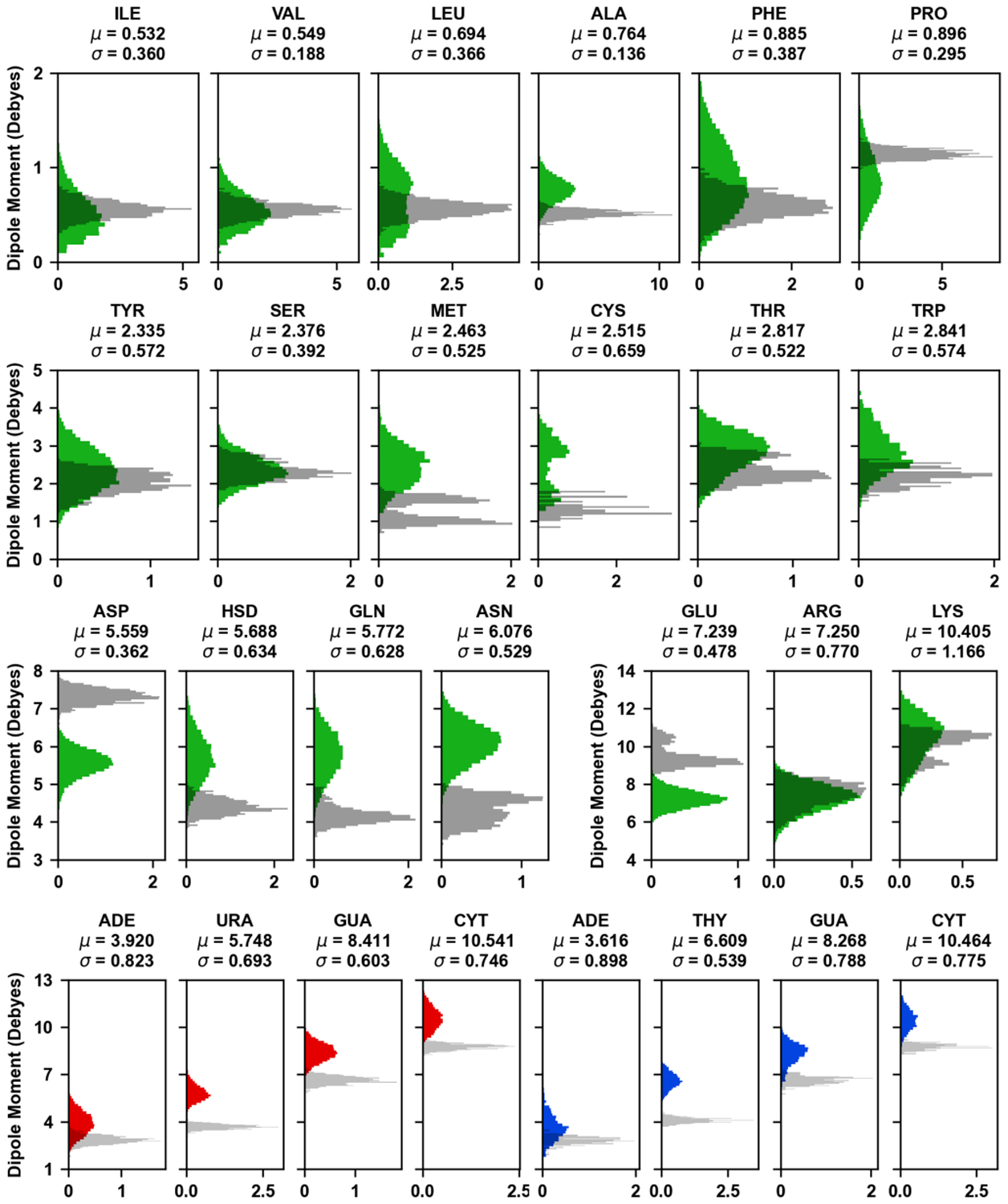
References
-
- Renaud J-P; Chari A; Ciferri C; Liu W.-t.; Rémigy H-W; Stark H; Wiesmann C, Cryo-EM in drug discovery: achievements, limitations and prospects. Nature Reviews Drug Discovery 2018, 17 (7), 471–492. - PubMed
-
- Penfold J; Wagner NJ, Editorial overview: Recent applications of x-ray and neutron scattering techniques in colloid and interfacial science, characterised by increasing diversity and complexity. Current Opinion in Colloid & Interface Science 2019, 42.
-
- Sotomayor M; Schulten K, Single-molecule experiments in vitro and in silico. Science 2007, 316 (5828), 1144–8. - PubMed
-
- van der Kamp MW; Mulholland AJ, Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52 (16), 2708–28. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
