

Analysis of long and short enhancers in melanoma cell states
- PMID: 34874265
- PMCID: PMC8691835
- DOI: 10.7554/eLife.71735
Analysis of long and short enhancers in melanoma cell states
Abstract
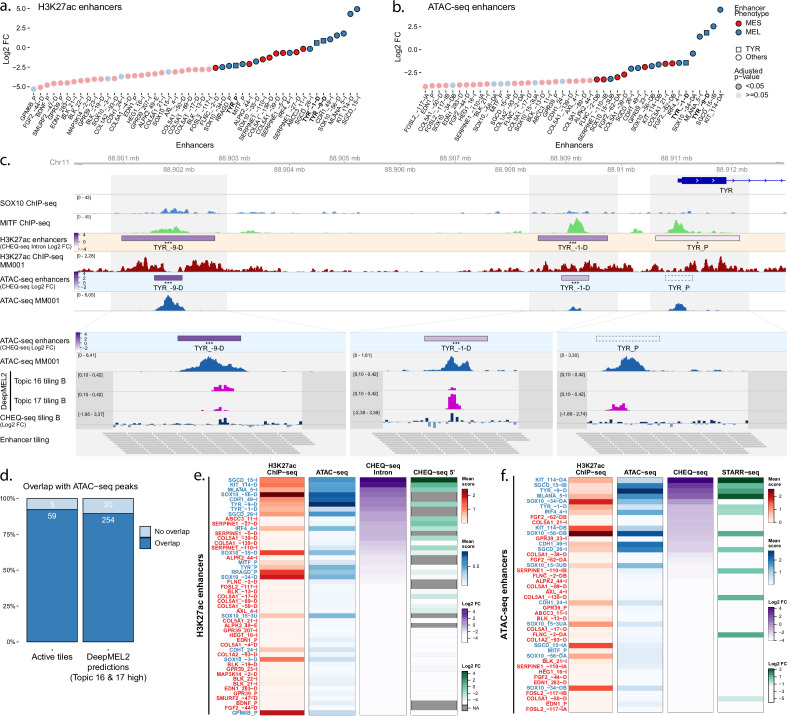
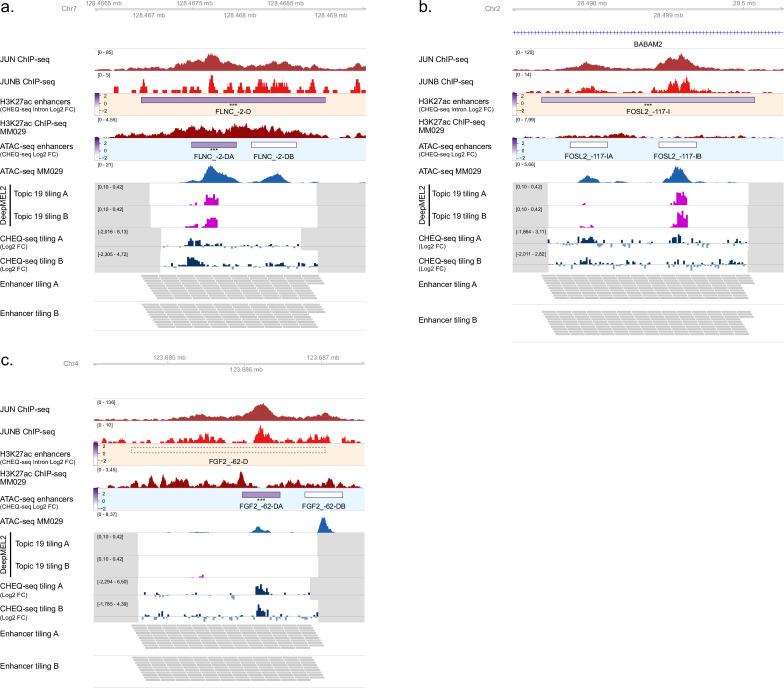
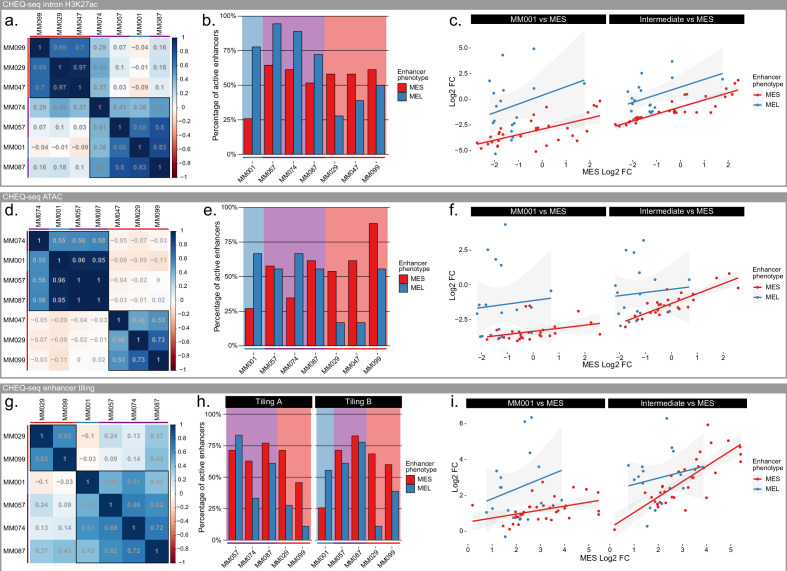
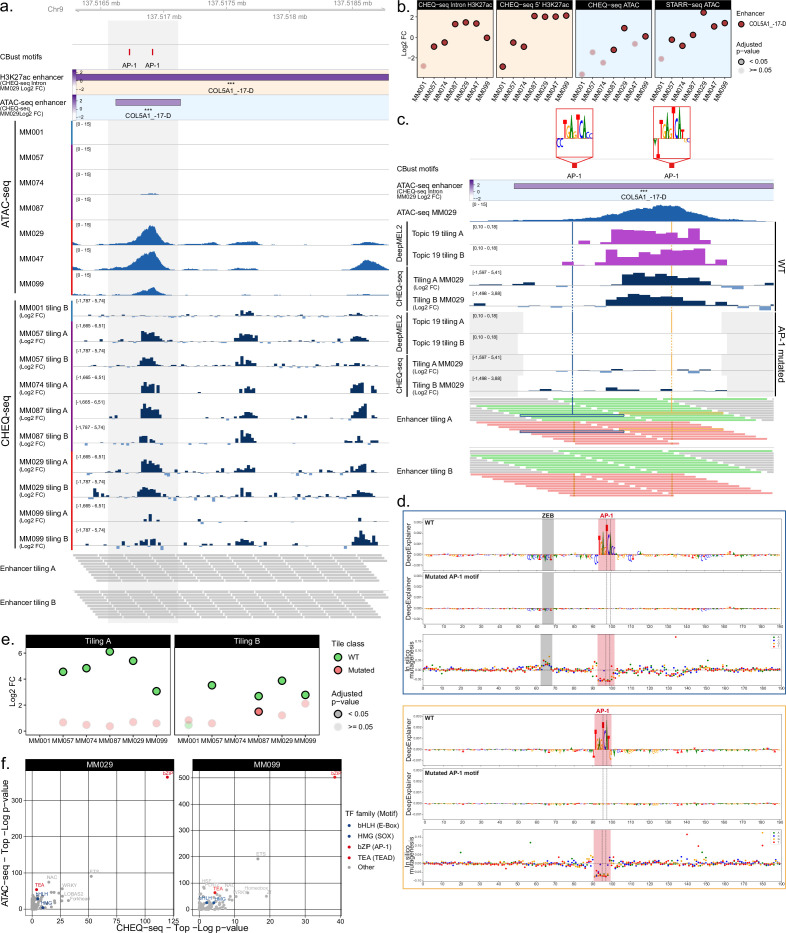
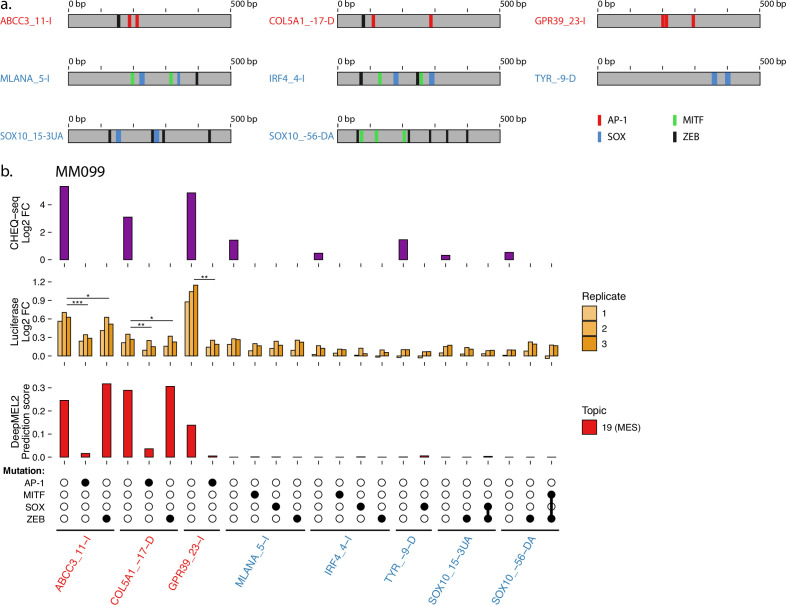
Understanding how enhancers drive cell-type specificity and efficiently identifying them is essential for the development of innovative therapeutic strategies. In melanoma, the melanocytic (MEL) and the mesenchymal-like (MES) states present themselves with different responses to therapy, making the identification of specific enhancers highly relevant. Using massively parallel reporter assays (MPRAs) in a panel of patient-derived melanoma lines (MM lines), we set to identify and decipher melanoma enhancers by first focusing on regions with state-specific H3K27 acetylation close to differentially expressed genes. An in-depth evaluation of those regions was then pursued by investigating the activity of overlapping ATAC-seq peaks along with a full tiling of the acetylated regions with 190 bp sequences. Activity was observed in more than 60% of the selected regions, and we were able to precisely locate the active enhancers within ATAC-seq peaks. Comparison of sequence content with activity, using the deep learning model DeepMEL2, revealed that AP-1 alone is responsible for the MES enhancer activity. In contrast, SOX10 and MITF both influence MEL enhancer function with SOX10 being required to achieve high levels of activity. Overall, our MPRAs shed light on the relationship between long and short sequences in terms of their sequence content, enhancer activity, and specificity across melanoma cell states.
Keywords: MPRA; chromosomes; enhancers; gene expression; genetics; genomics; human; melanoma.
© 2021, Mauduit et al.
Conflict of interest statement
DM, IT, LM, Md, VC, GH, JD, JW, SA No competing interests declared
Figures



















References
-
- Atak ZK, Taskiran II, Demeulemeester J, Flerin C, Mauduit D, Minnoye L, Hulselmans G, Christiaens V, Ghanem GE, Wouters J, Aerts S. Interpretation of allele-specific chromatin accessibility using cell state-aware deep learning. Genome Research. 2021;31:1082–1096. doi: 10.1101/gr.260851.120. - DOI - PMC - PubMed
-
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. PNAS. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
