

Population Histories and Genomic Diversity of South American Natives
- PMID: 34875092
- PMCID: PMC8789086
- DOI: 10.1093/molbev/msab339
Population Histories and Genomic Diversity of South American Natives
Abstract
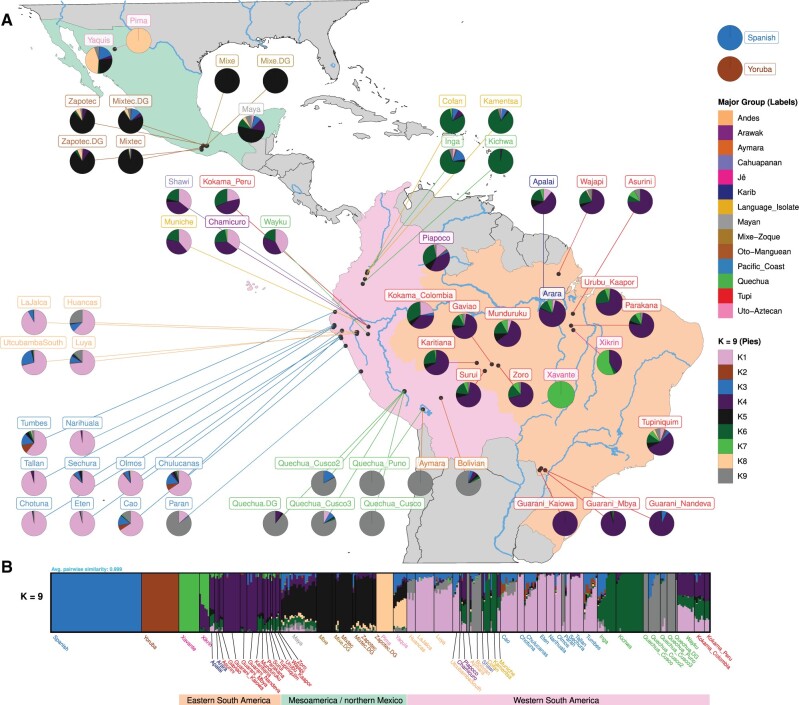
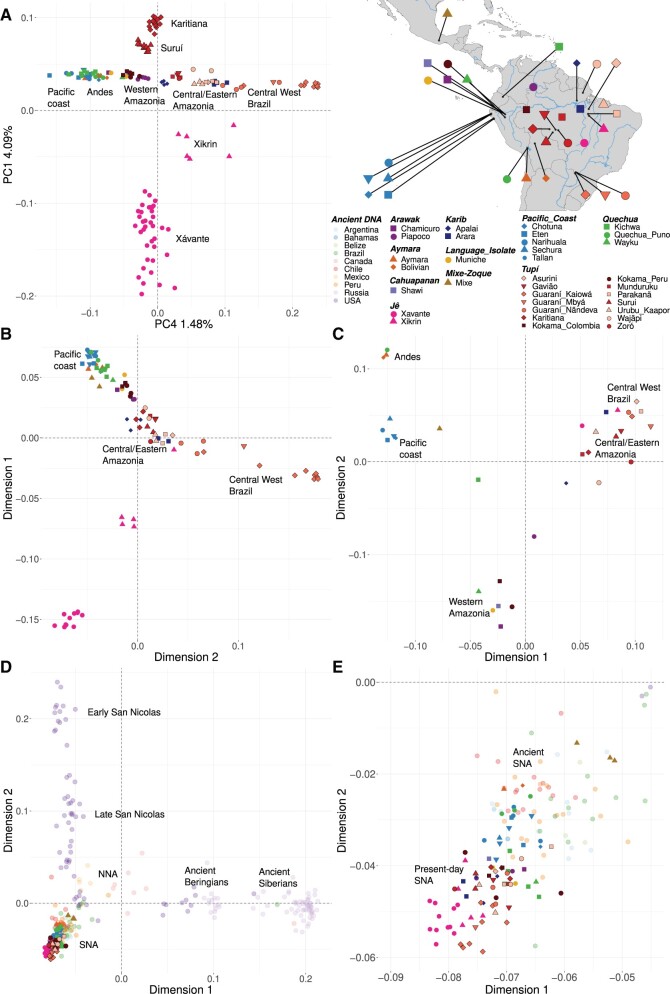
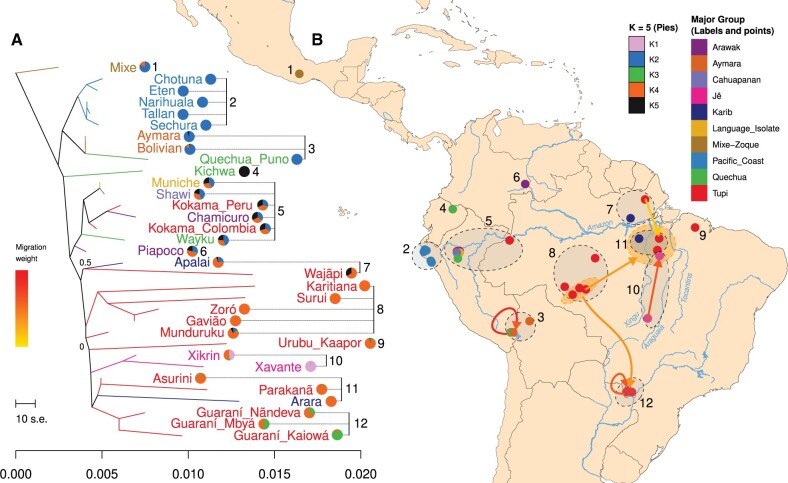
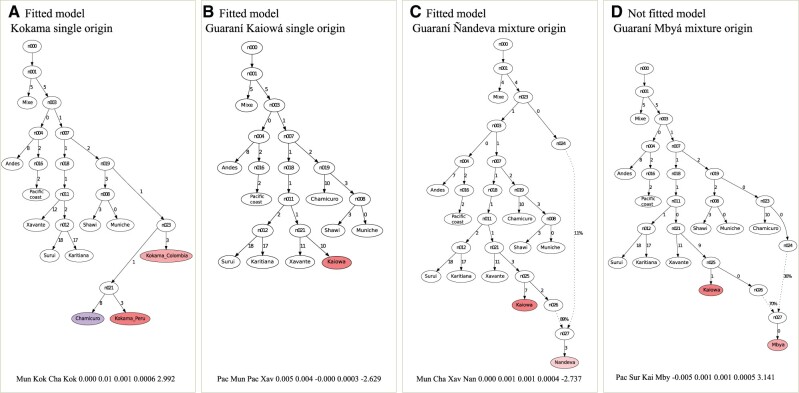
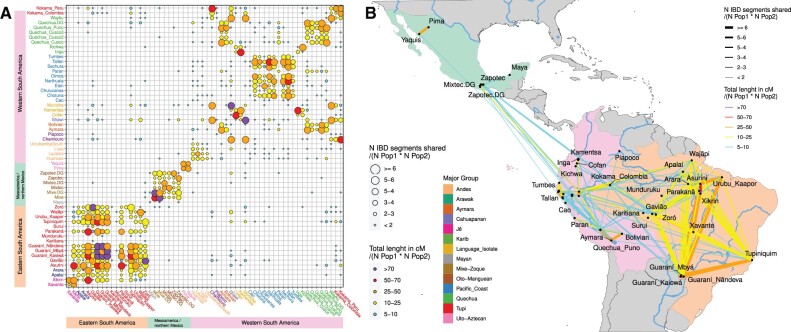
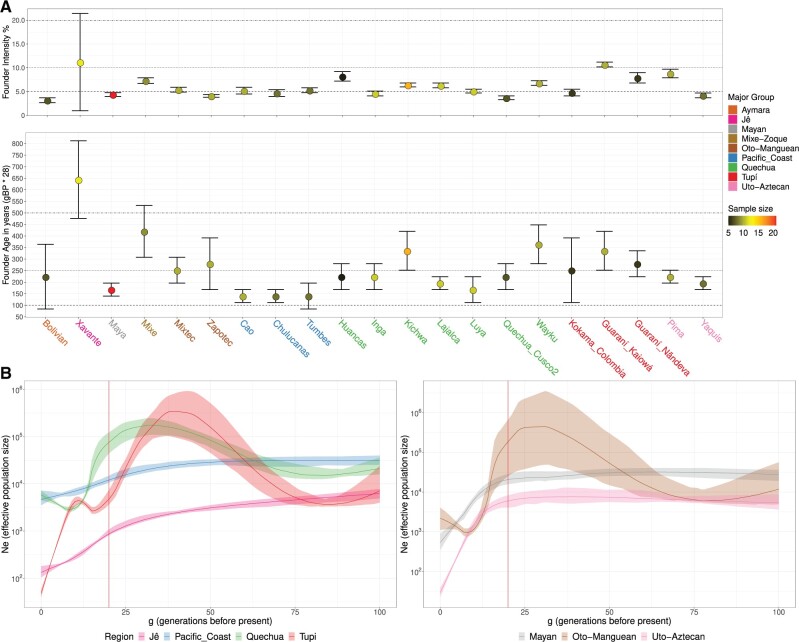
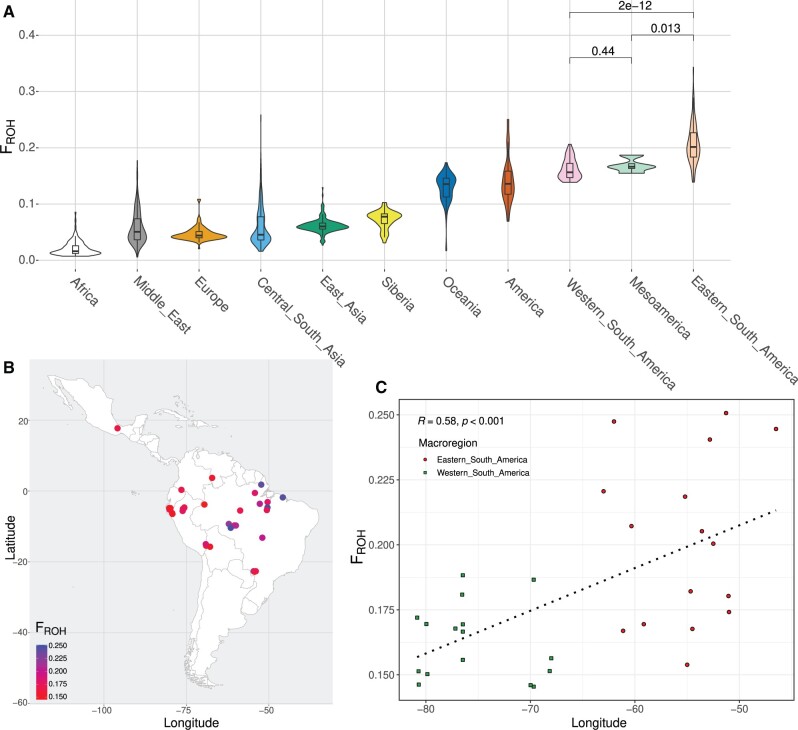
South America is home to one of the most culturally diverse present-day native populations. However, the dispersion pattern, genetic substructure, and demographic complexity within South America are still poorly understood. Based on genome-wide data of 58 native populations, we provide a comprehensive scenario of South American indigenous groups considering the genomic, environmental, and linguistic data. Clear patterns of genetic structure were inferred among the South American natives, presenting at least four primary genetic clusters in the Amazonian and savanna regions and three clusters in the Andes and Pacific coast. We detected a cline of genetic variation along a west-east axis, contradicting a hard Andes-Amazon divide. This longitudinal genetic variation seemed to have been shaped by both serial population bottlenecks and isolation by distance. Results indicated that present-day South American substructures recapitulate ancient macroregional ancestries and western Amazonia groups show genetic evidence of cultural exchanges that led to language replacement in precontact times. Finally, demographic inferences pointed to a higher resilience of the western South American groups regarding population collapses caused by the European invasion and indicated precontact population reductions and demic expansions in South America.
Keywords: Andes-Amazonia divide; genetics; native Americans; settlement of South America.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Adhikari K, Chacón-Duque JC, Mendoza-Revilla J, Fuentes-Guajardo M, Ruiz-Linares A.. 2017. The genetic diversity of the Americas. Annu Rev Genomics Hum Genet. 18:277–296. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
