

Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation
- PMID: 34876685
- PMCID: PMC8683376
- DOI: 10.1038/s41556-021-00774-y
Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation
Erratum in
-
Author Correction: Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation.Nat Cell Biol. 2022 Jan;24(1):123. doi: 10.1038/s41556-021-00833-4. Nat Cell Biol. 2022. PMID: 34983954 Free PMC article. No abstract available.
Abstract
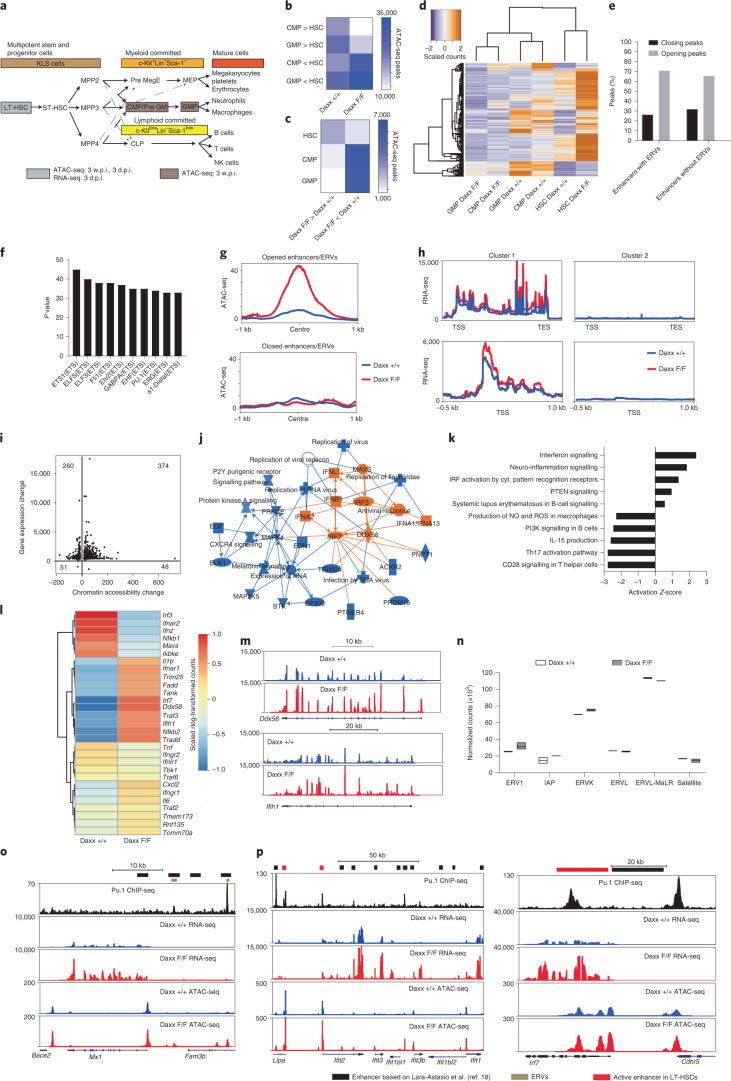
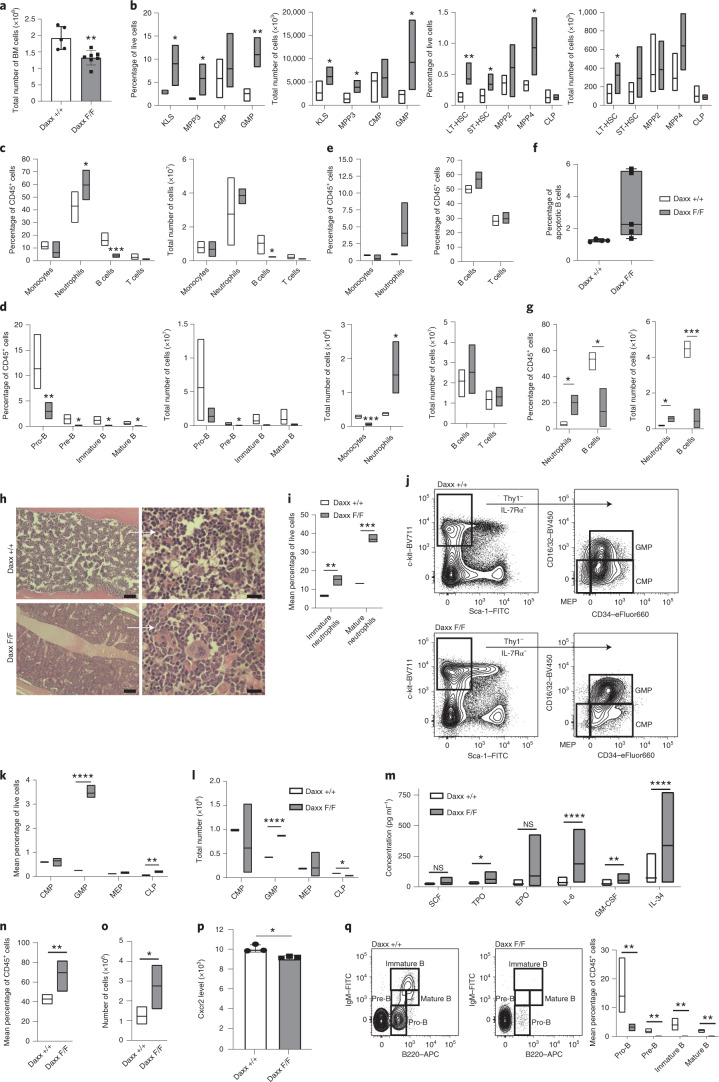
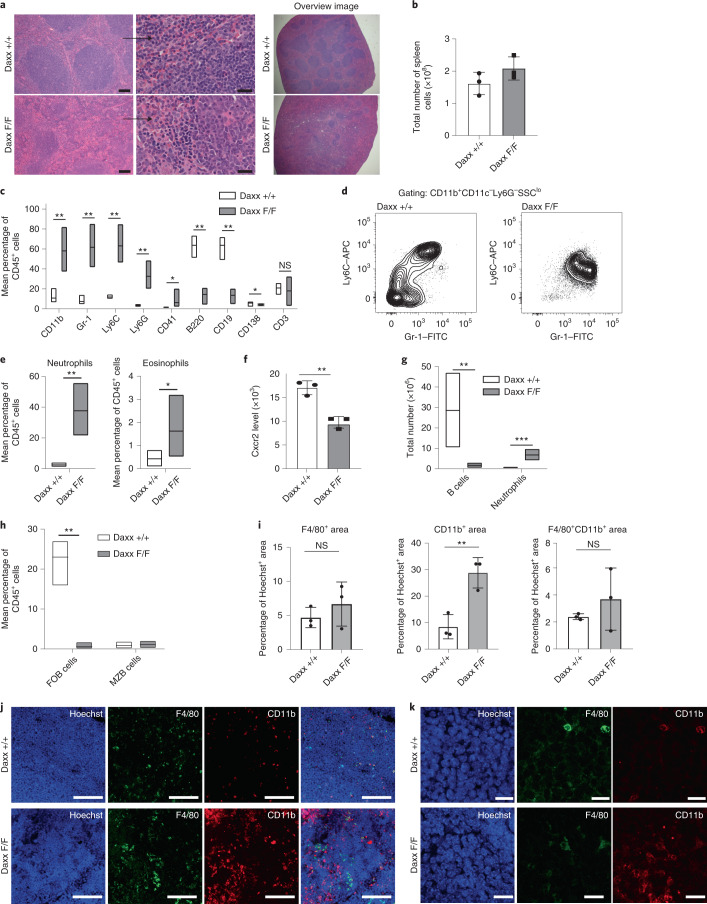
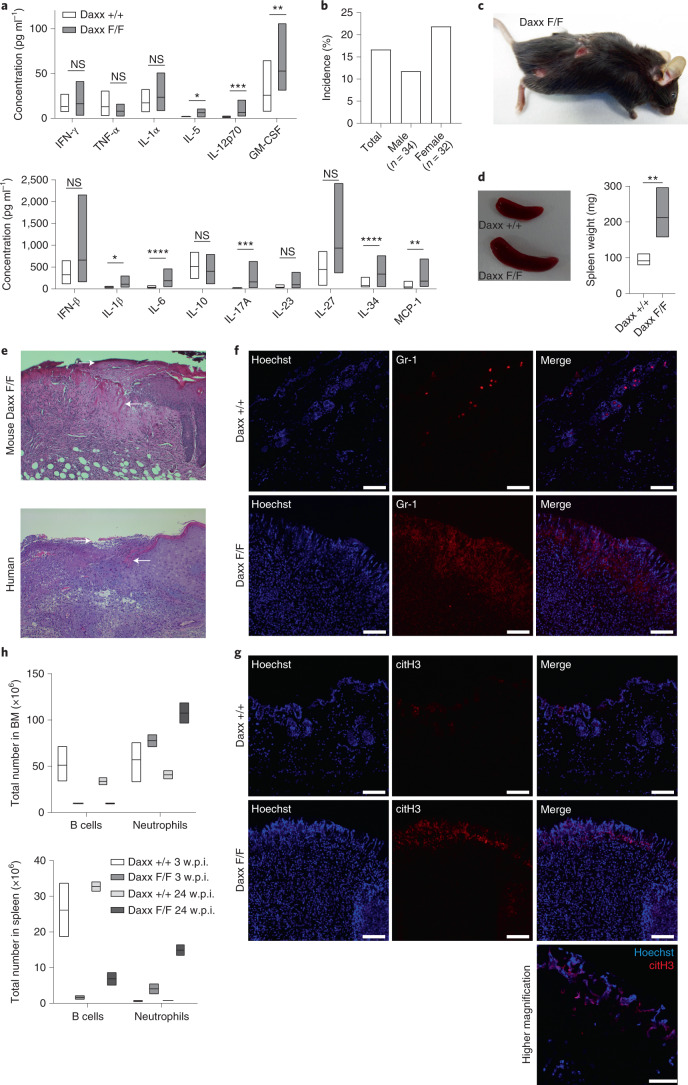
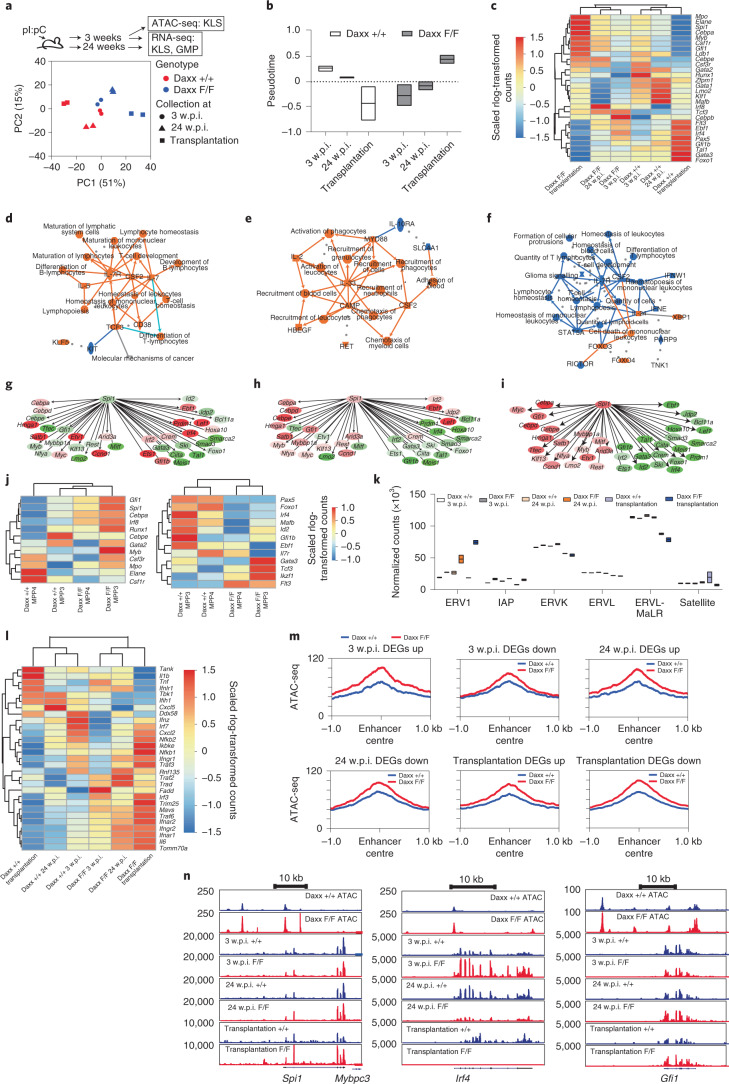
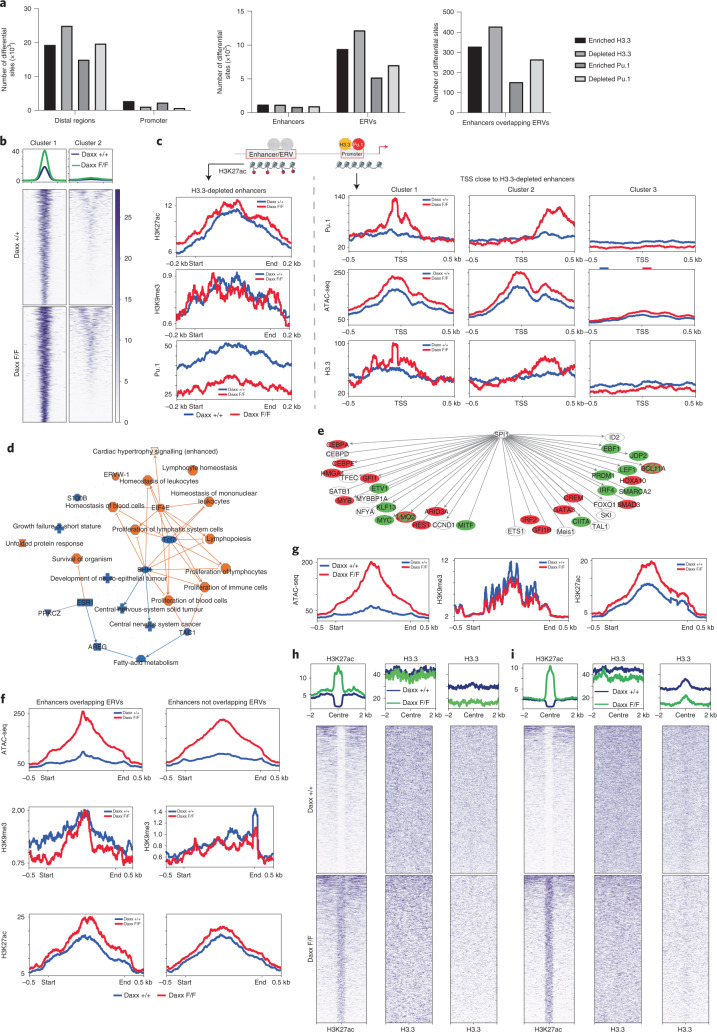
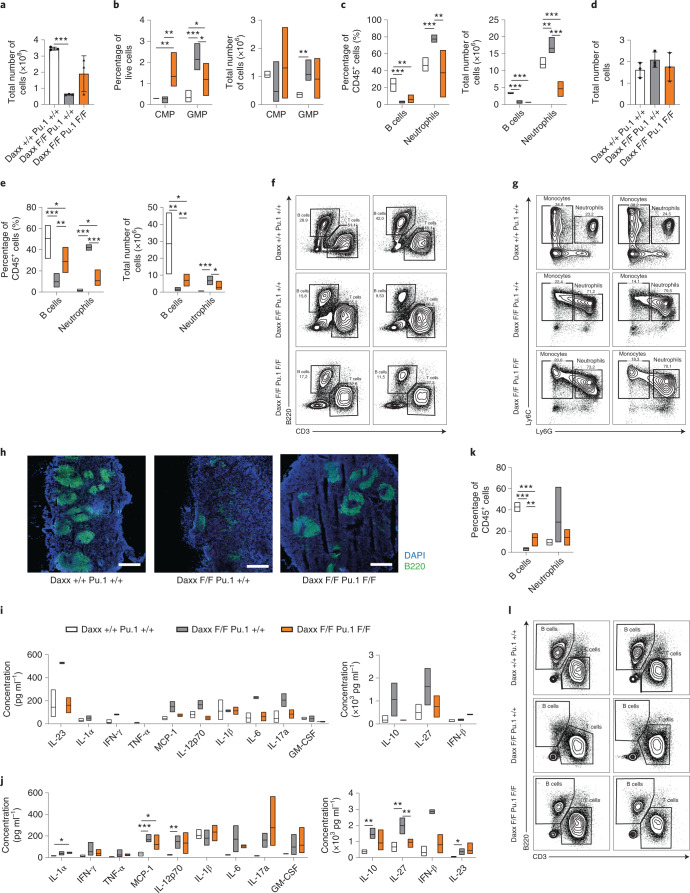
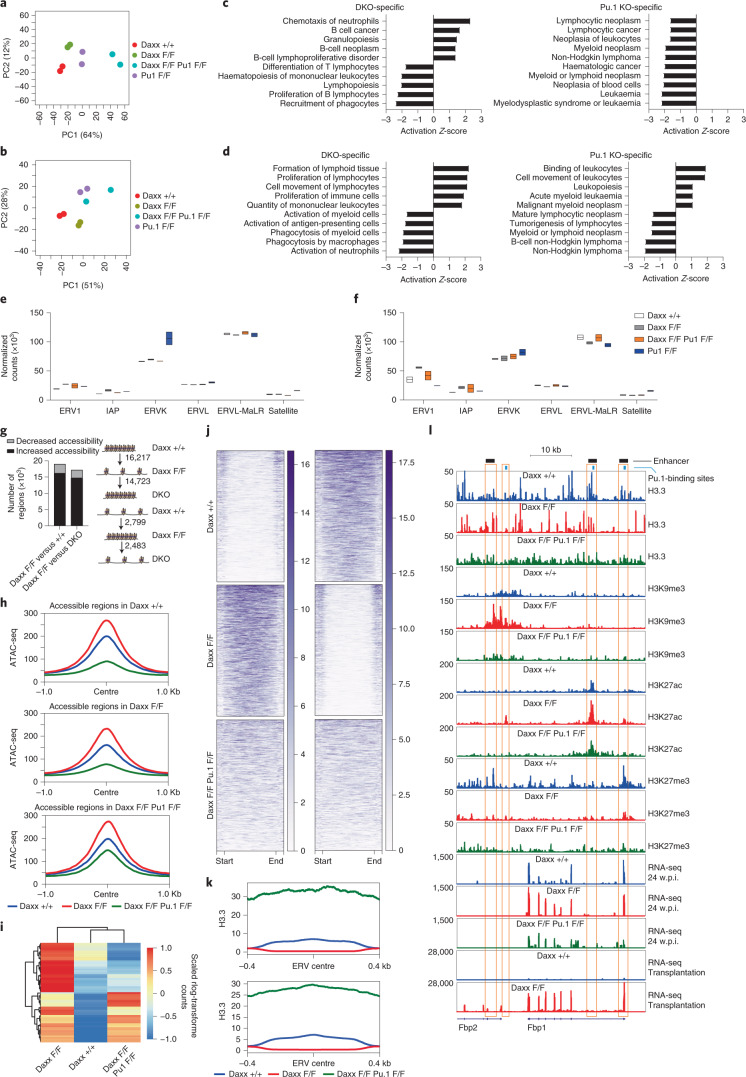
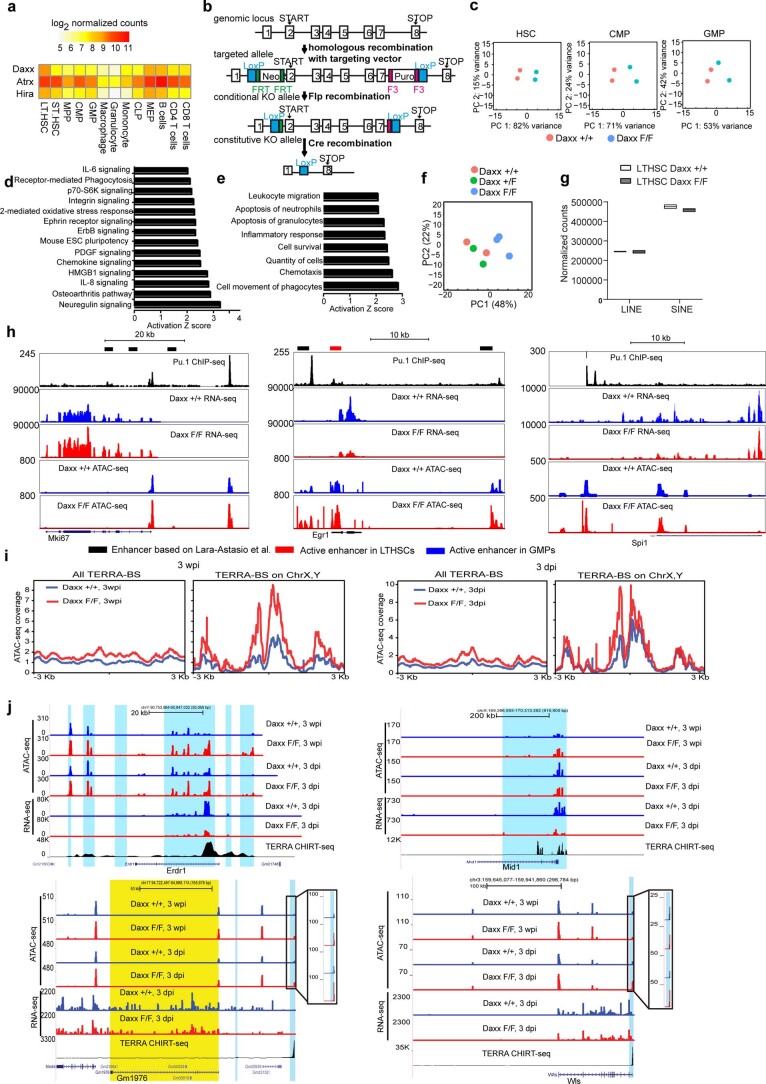
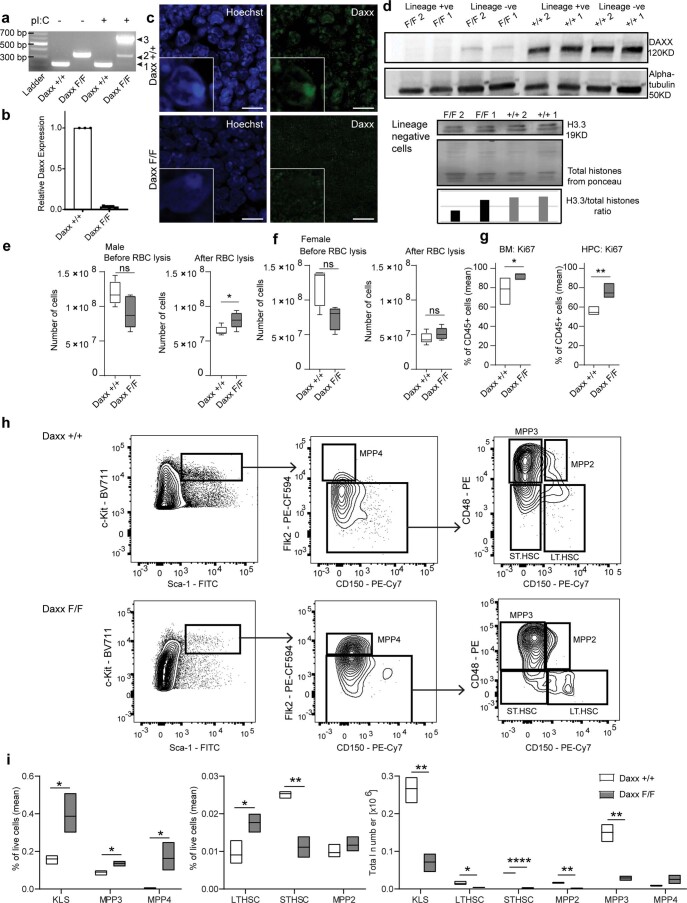
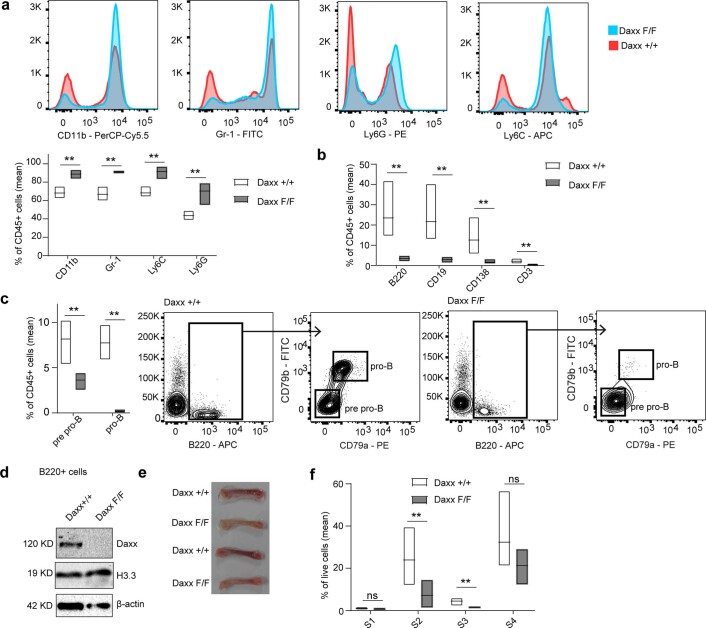
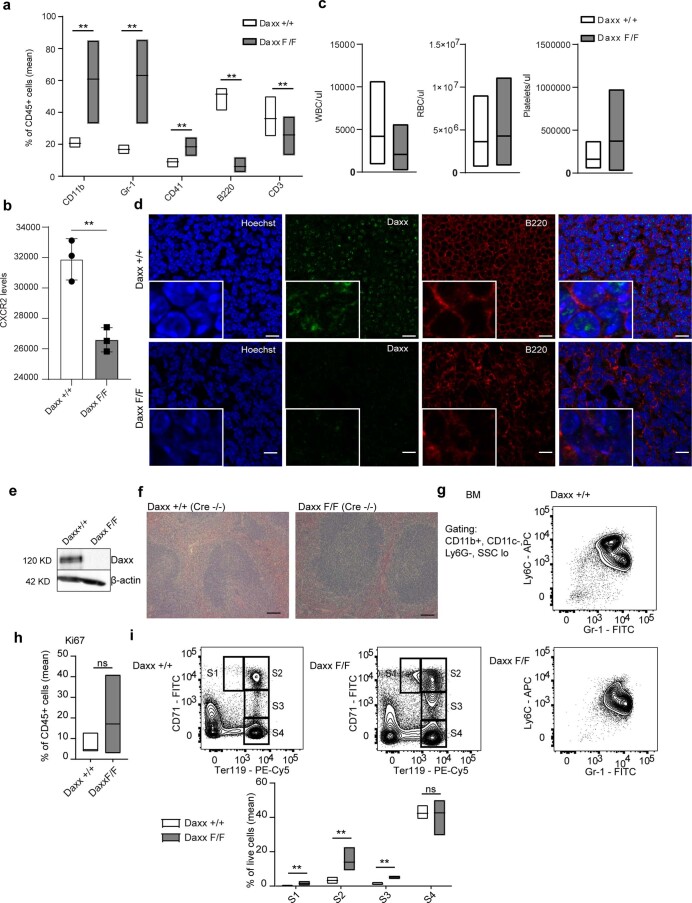
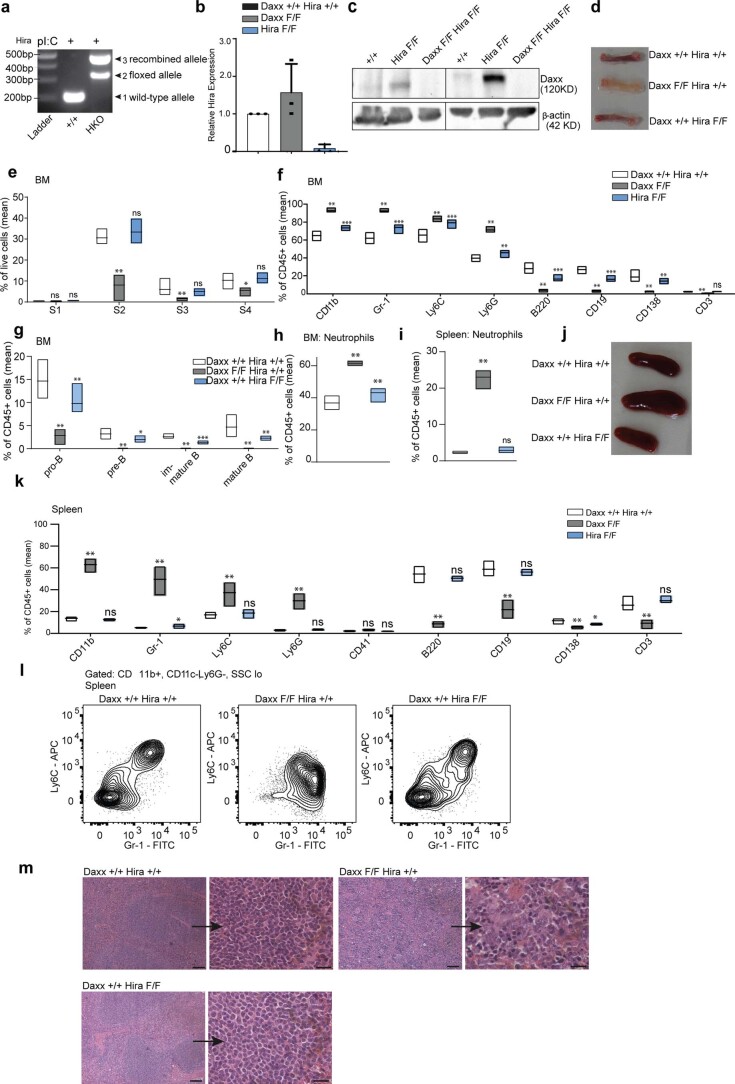
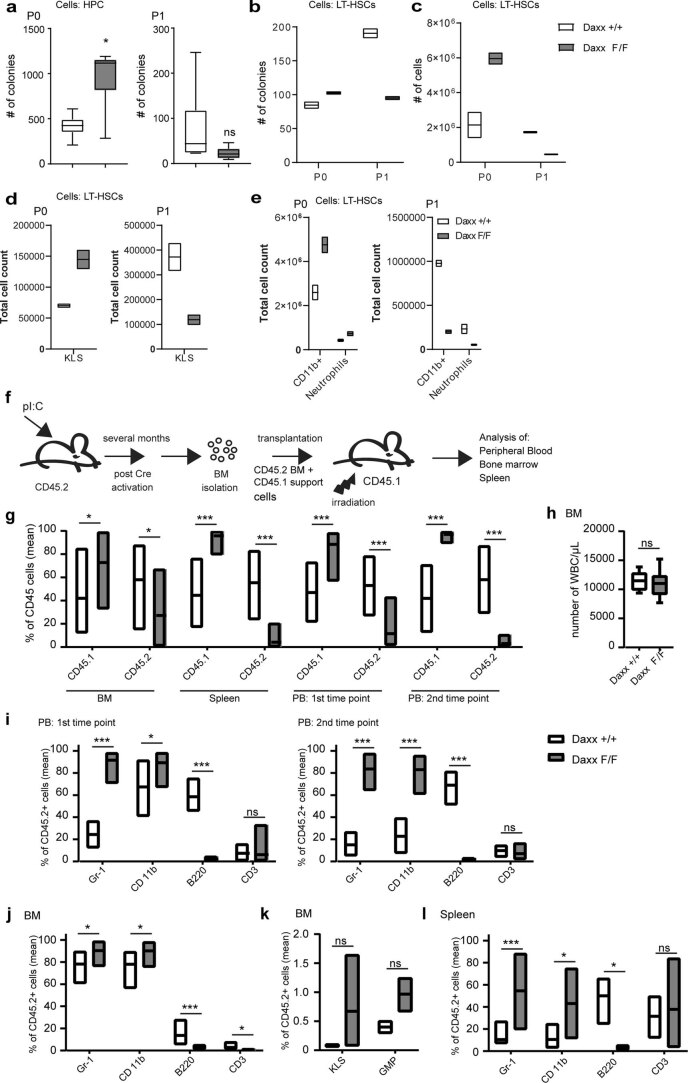
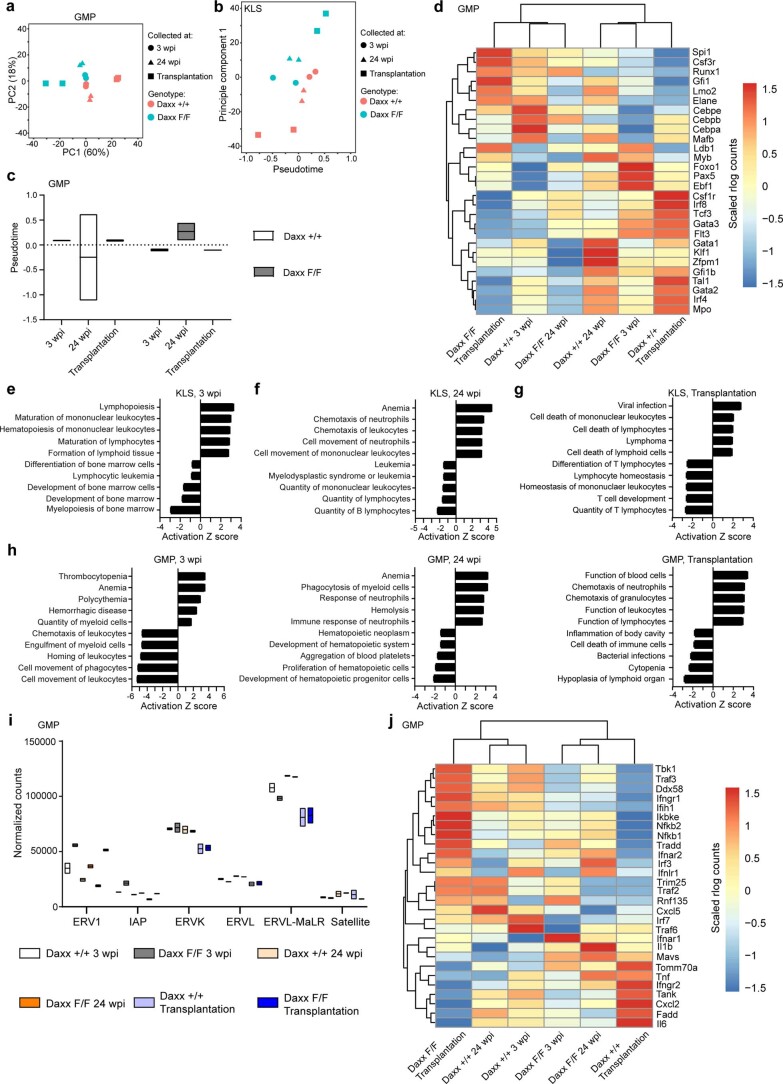
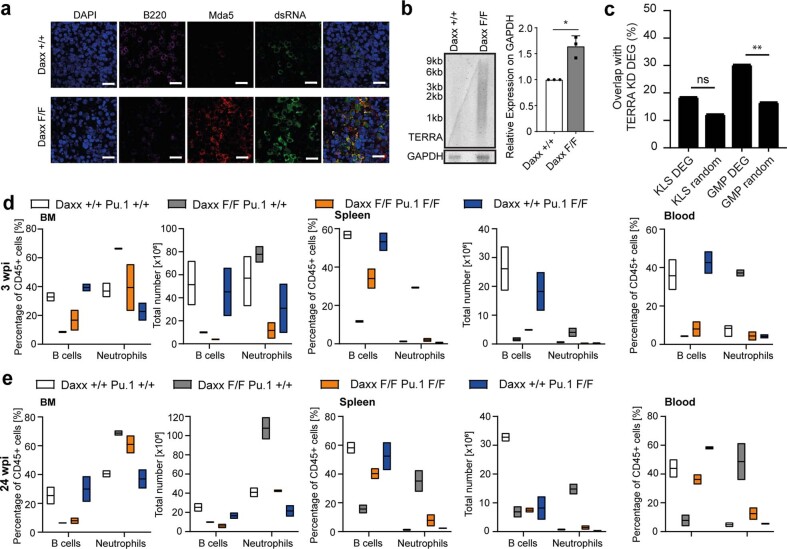
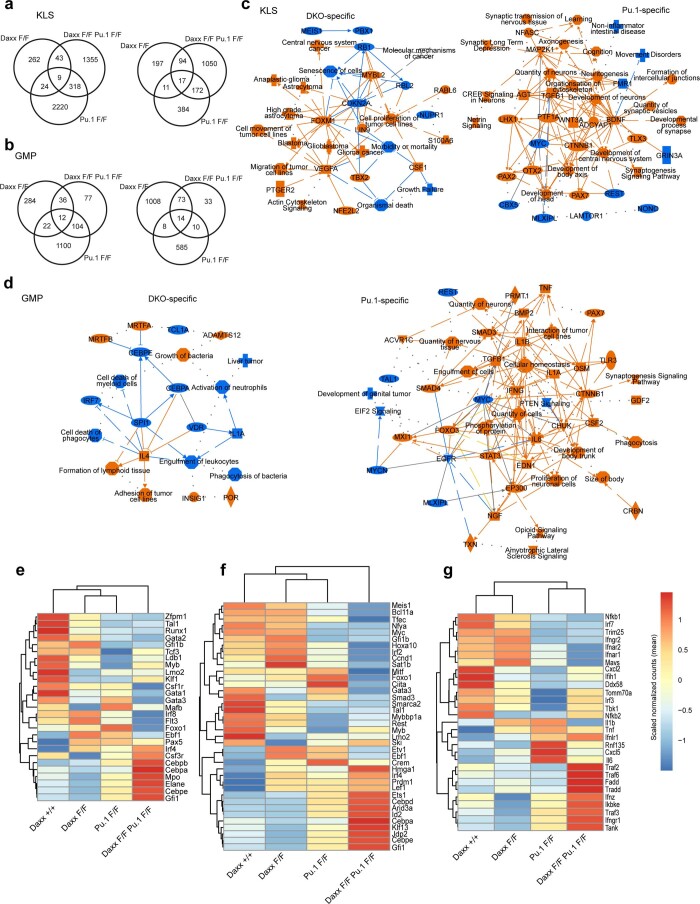
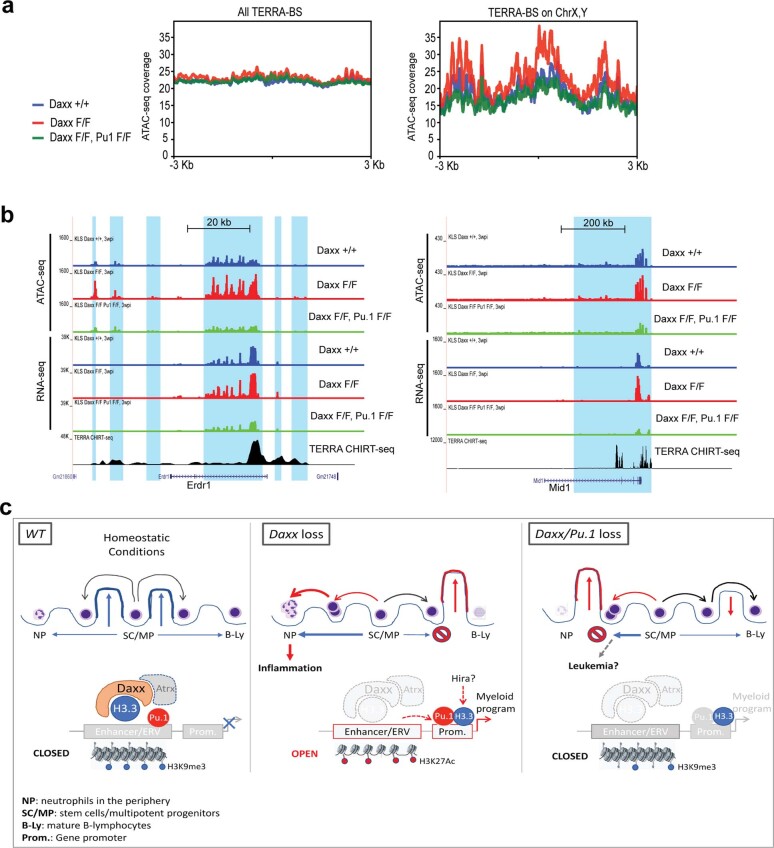
Defective silencing of retrotransposable elements has been linked to inflammageing, cancer and autoimmune diseases. However, the underlying mechanisms are only partially understood. Here we implicate the histone H3.3 chaperone Daxx, a retrotransposable element repressor inactivated in myeloid leukaemia and other neoplasms, in protection from inflammatory disease. Loss of Daxx alters the chromatin landscape, H3.3 distribution and histone marks of haematopoietic progenitors, leading to engagement of a Pu.1-dependent transcriptional programme for myelopoiesis at the expense of B-cell differentiation. This causes neutrophilia and inflammation, predisposing mice to develop an autoinflammatory skin disease. While these molecular and phenotypic perturbations are in part reverted in animals lacking both Pu.1 and Daxx, haematopoietic progenitors in these mice show unique chromatin and transcriptome alterations, suggesting an interaction between these two pathways. Overall, our findings implicate retrotransposable element silencing in haematopoiesis and suggest a cross-talk between the H3.3 loading machinery and the pioneer transcription factor Pu.1.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
H3.3 safeguards haematopoietic ERV-quilibrium.Nat Cell Biol. 2022 Jan;24(1):7-9. doi: 10.1038/s41556-021-00758-y. Nat Cell Biol. 2022. PMID: 34961795 No abstract available.
References
-
- Kunarso G, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010;42:631–634. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
