

A single-cell transcriptomic landscape of the lungs of patients with COVID-19
- PMID: 34876692
- PMCID: PMC8650955
- DOI: 10.1038/s41556-021-00796-6
A single-cell transcriptomic landscape of the lungs of patients with COVID-19
Abstract
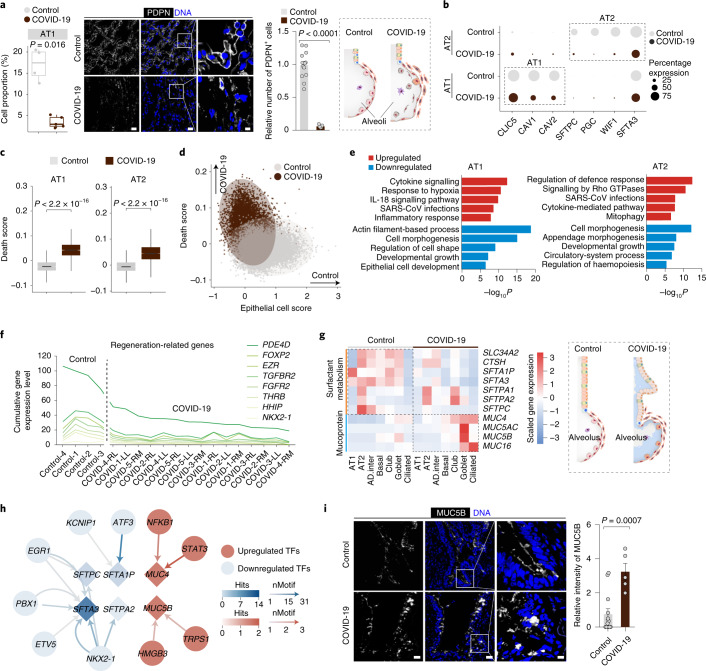
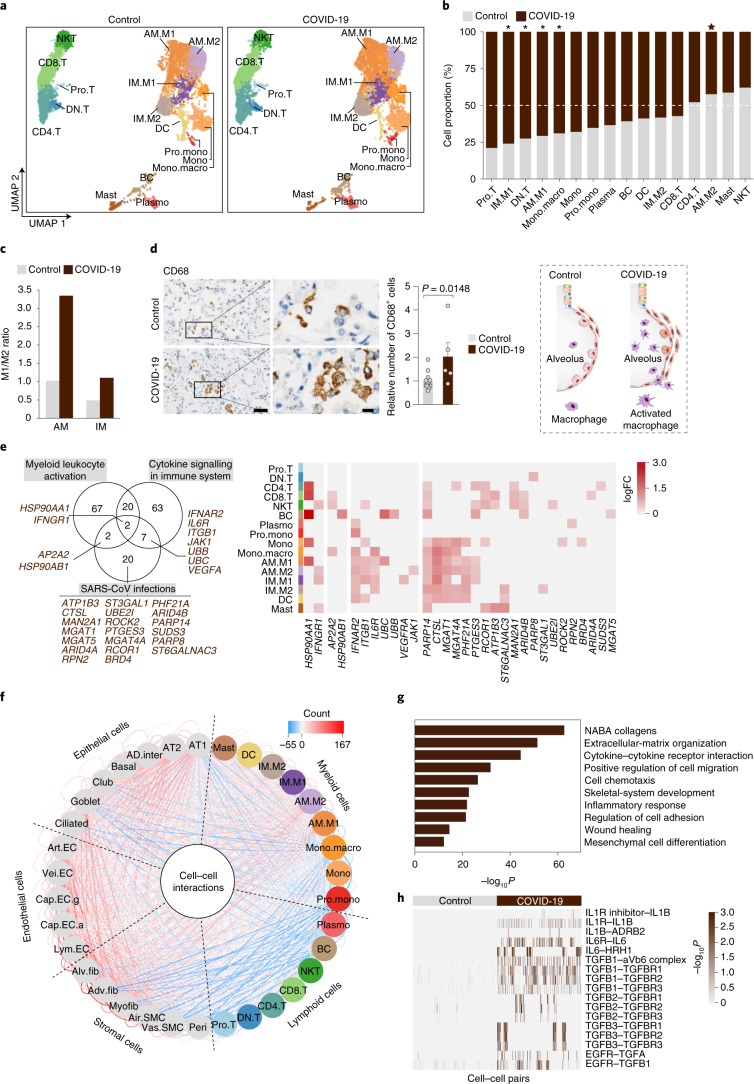
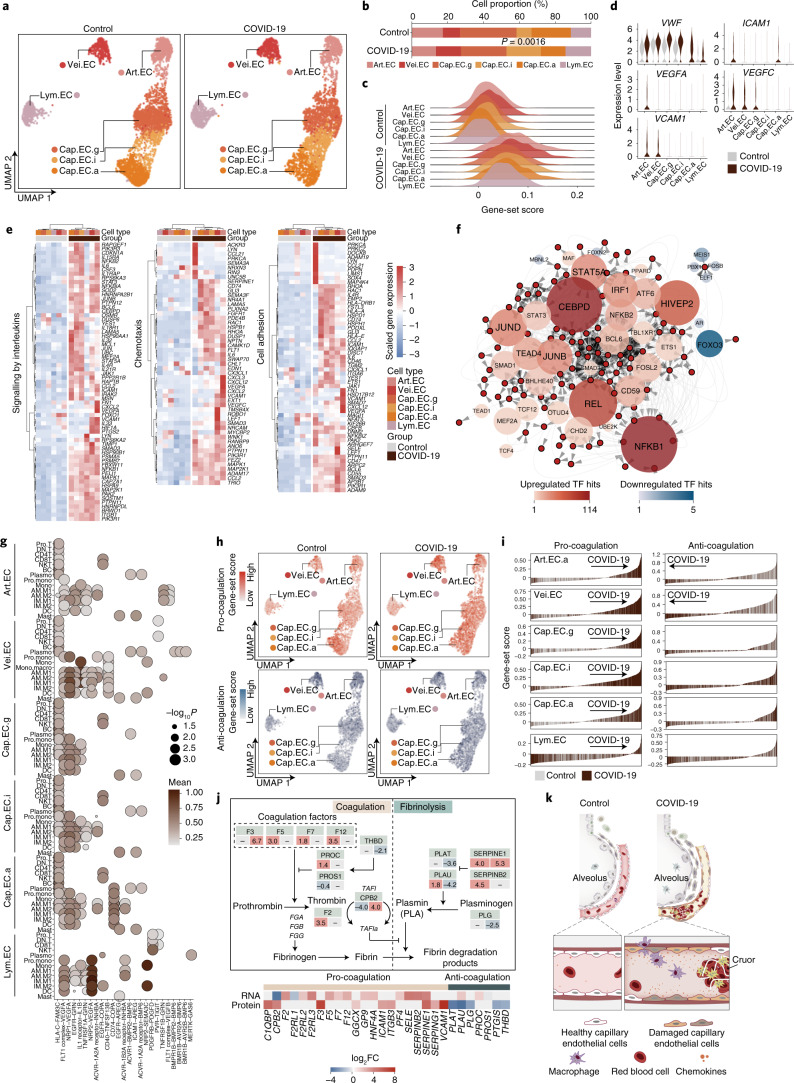
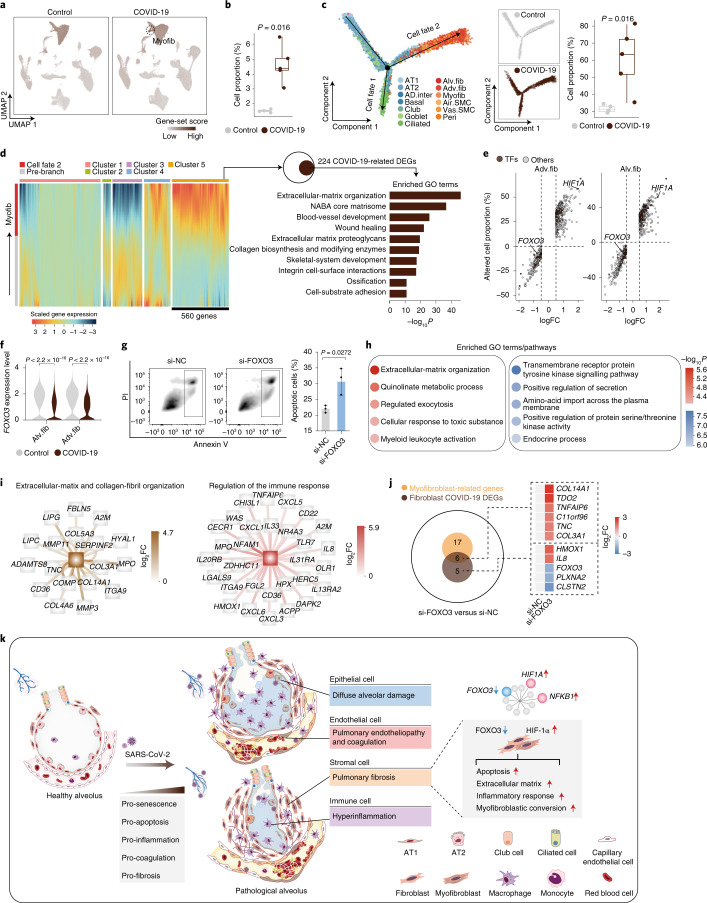
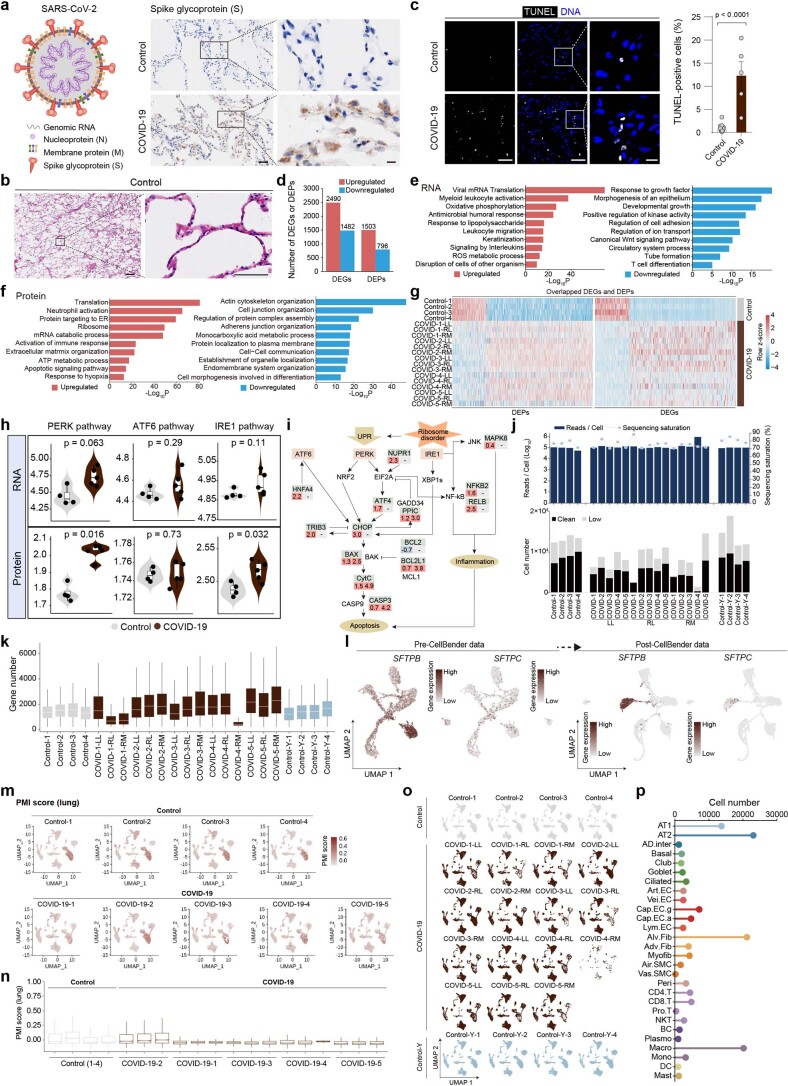
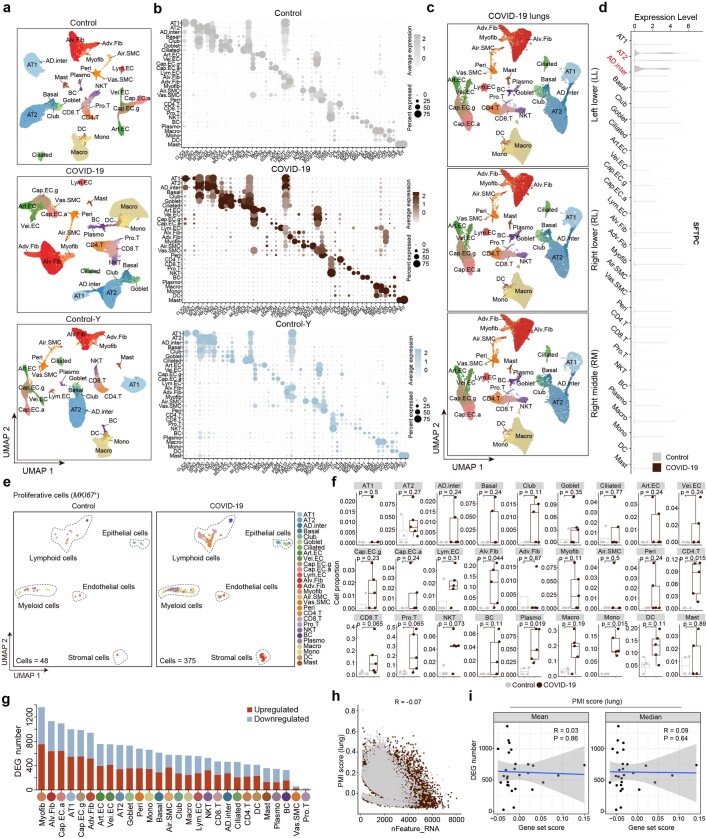
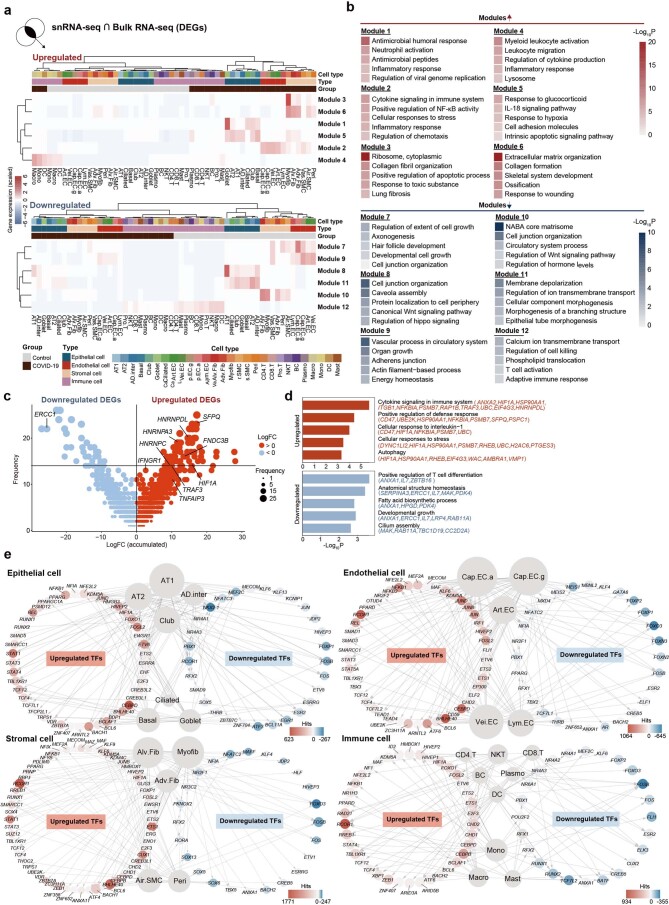
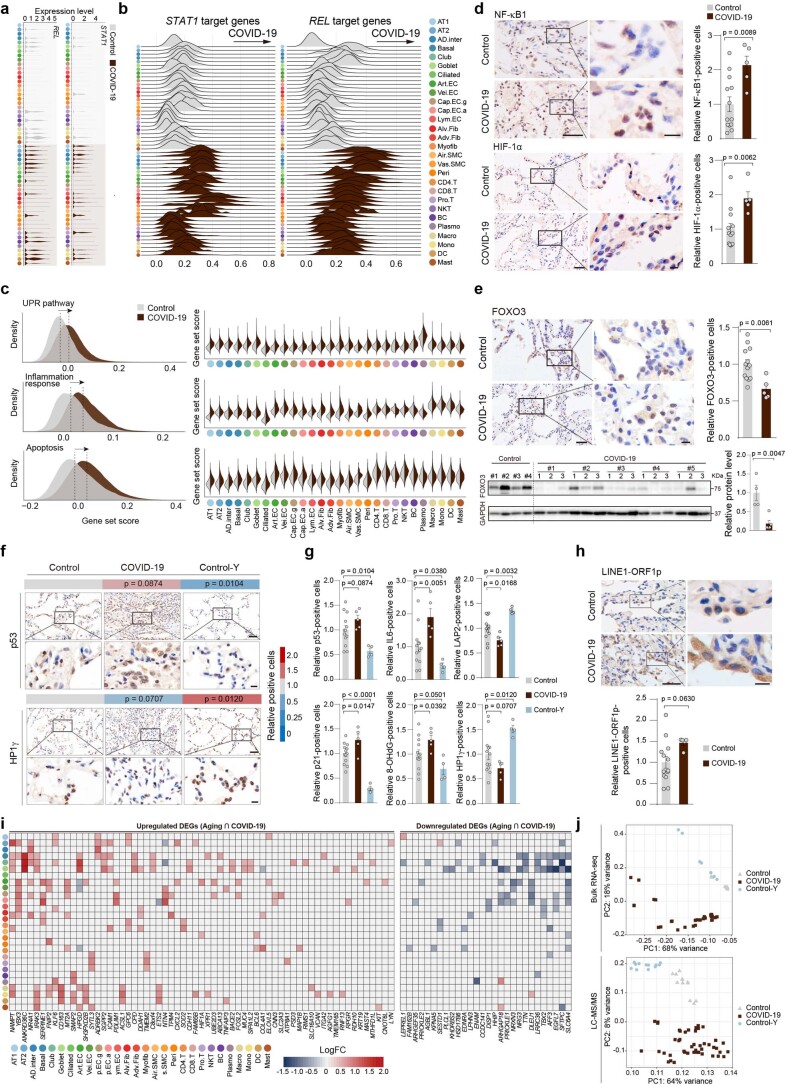
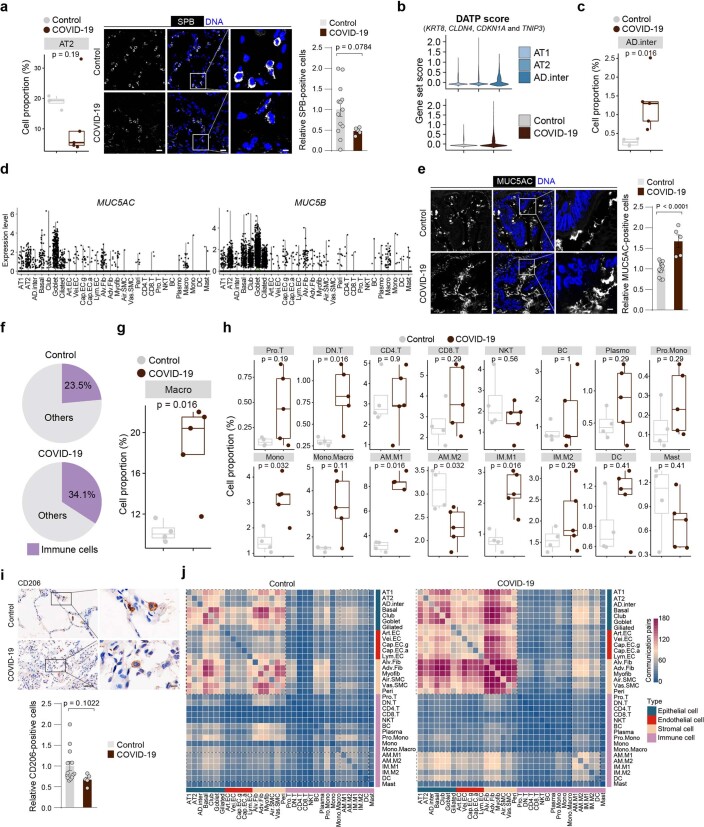
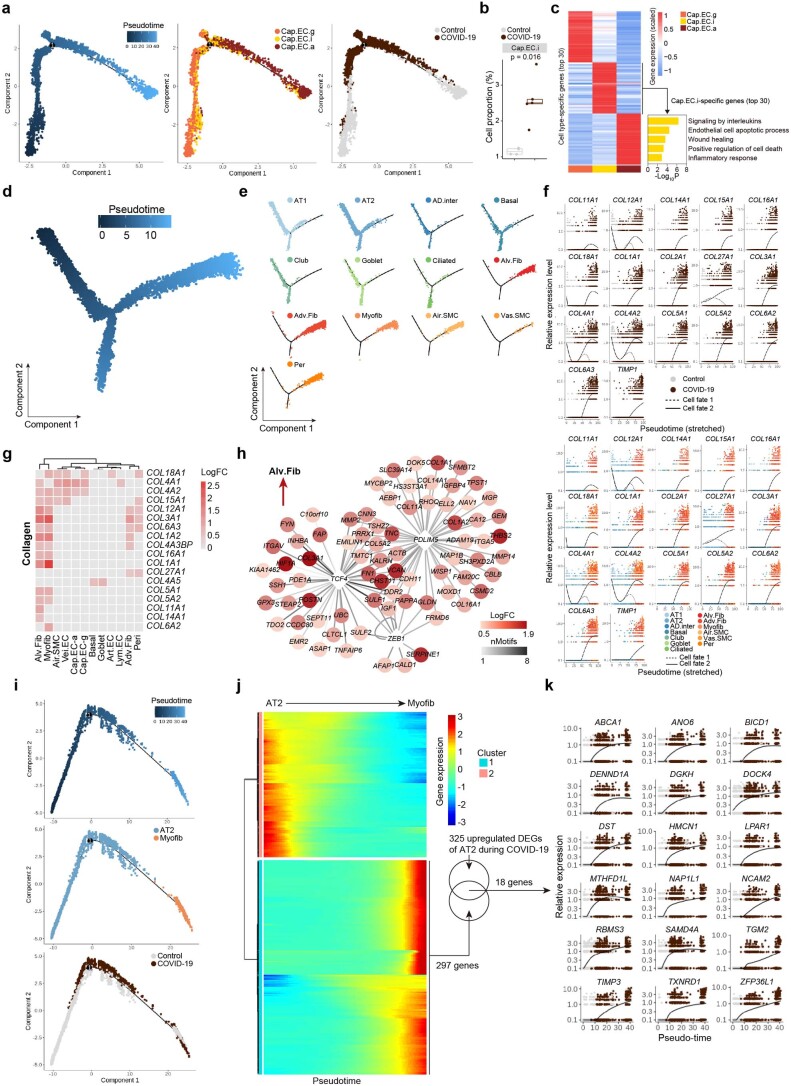
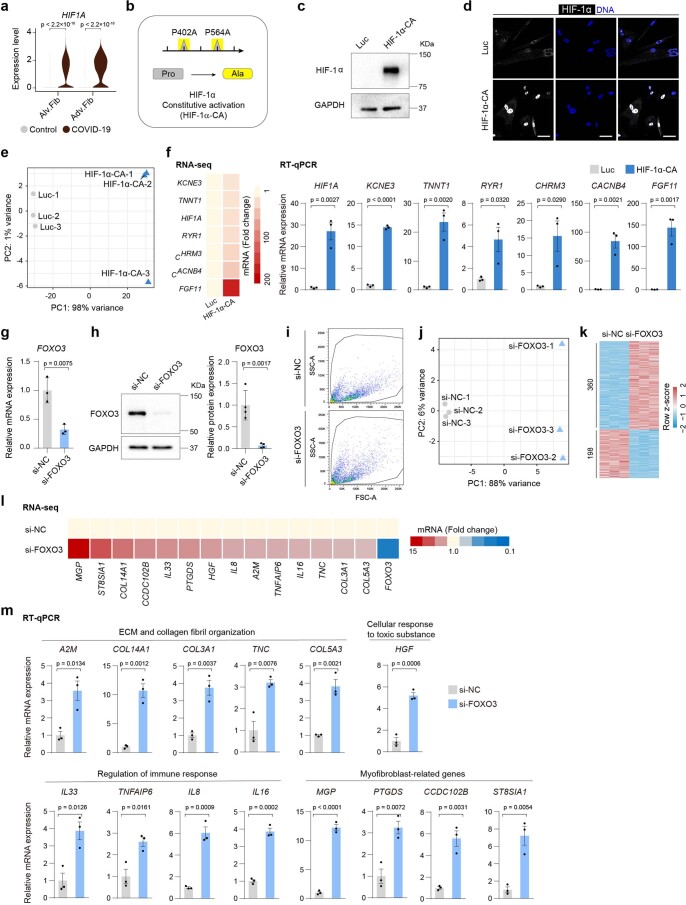
The lung is the primary organ targeted by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), making respiratory failure a leading coronavirus disease 2019 (COVID-19)-related mortality. However, our cellular and molecular understanding of how SARS-CoV-2 infection drives lung pathology is limited. Here we constructed multi-omics and single-nucleus transcriptomic atlases of the lungs of patients with COVID-19, which integrate histological, transcriptomic and proteomic analyses. Our work reveals the molecular basis of pathological hallmarks associated with SARS-CoV-2 infection in different lung and infiltrating immune cell populations. We report molecular fingerprints of hyperinflammation, alveolar epithelial cell exhaustion, vascular changes and fibrosis, and identify parenchymal lung senescence as a molecular state of COVID-19 pathology. Moreover, our data suggest that FOXO3A suppression is a potential mechanism underlying the fibroblast-to-myofibroblast transition associated with COVID-19 pulmonary fibrosis. Our work depicts a comprehensive cellular and molecular atlas of the lungs of patients with COVID-19 and provides insights into SARS-CoV-2-related pulmonary injury, facilitating the identification of biomarkers and development of symptomatic treatments.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
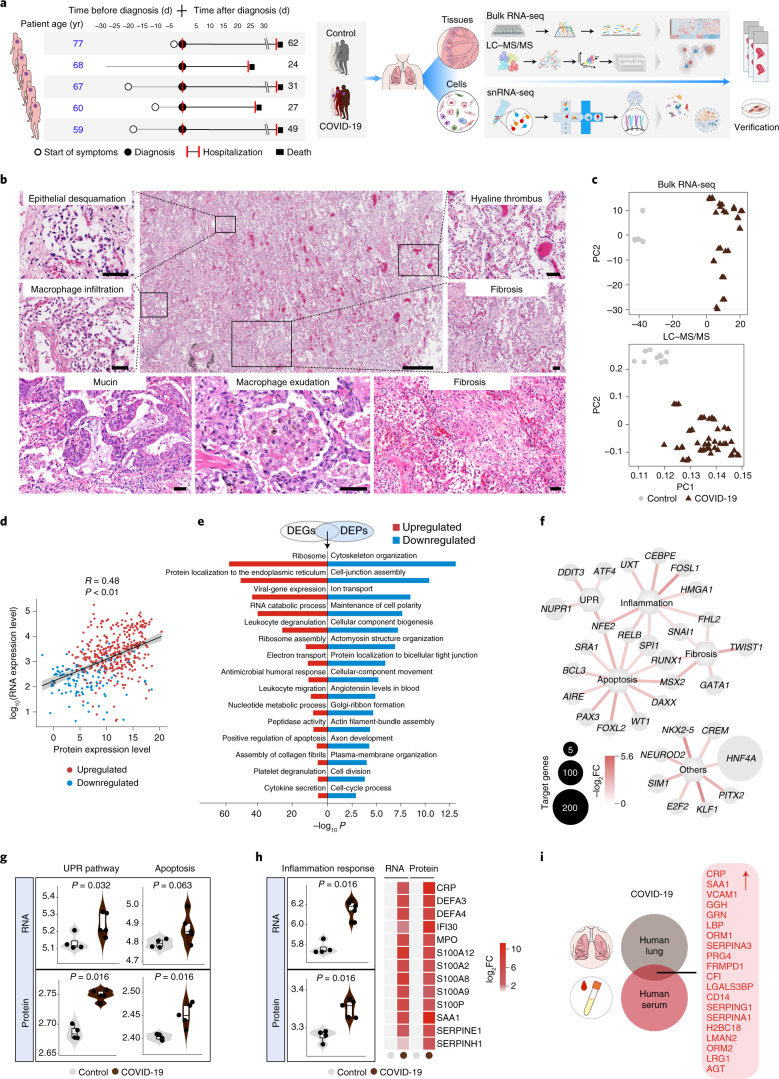
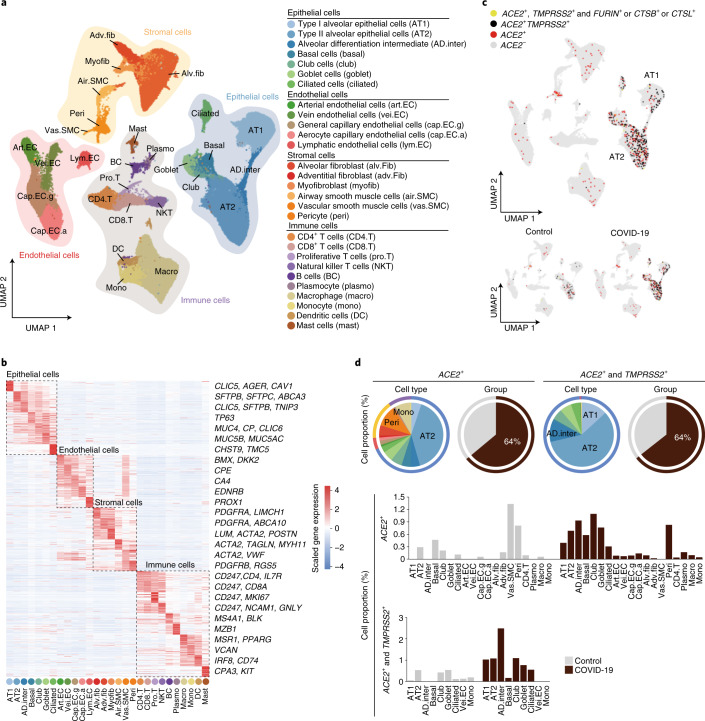
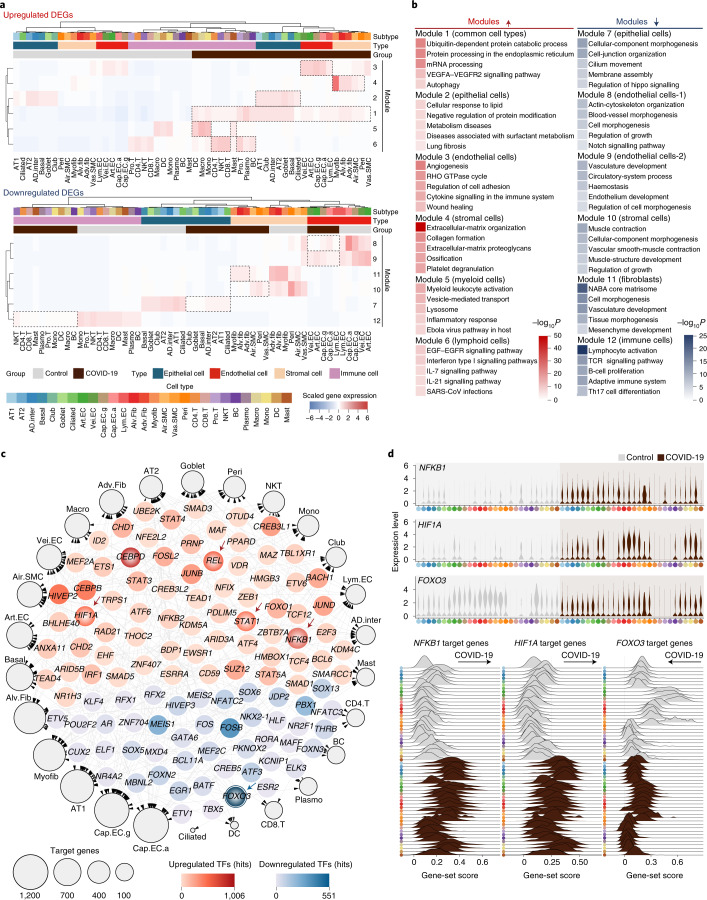
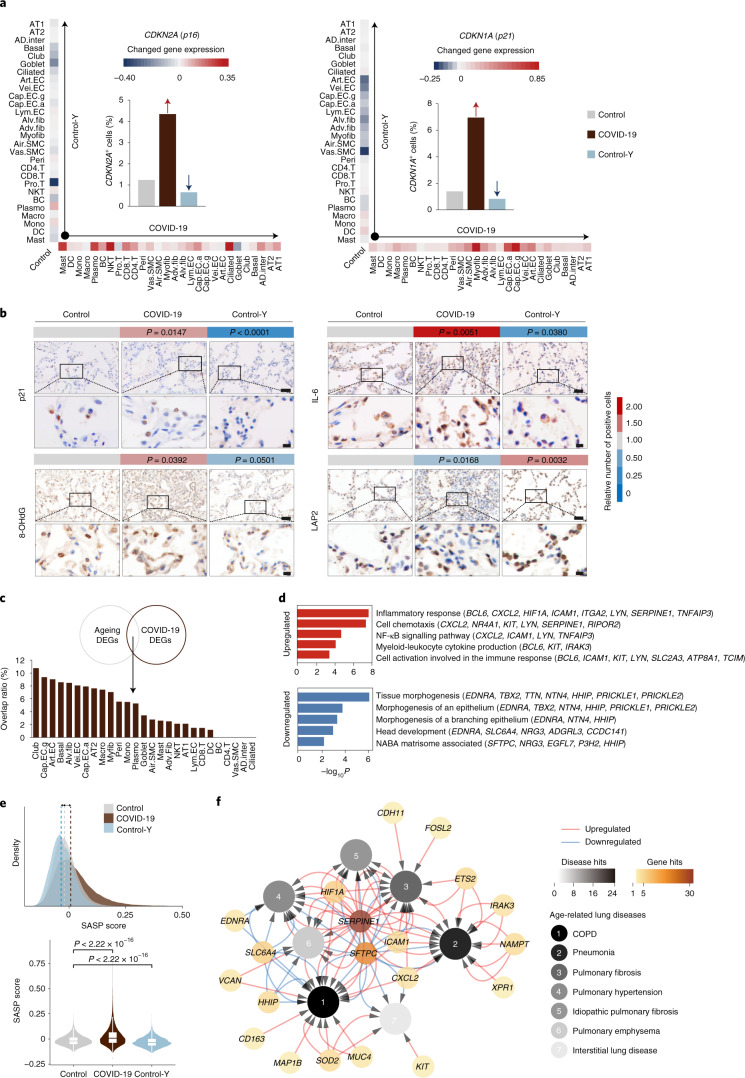

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
