

Malaria protection due to sickle haemoglobin depends on parasite genotype
- PMID: 34883497
- PMCID: PMC8810385
- DOI: 10.1038/s41586-021-04288-3
Malaria protection due to sickle haemoglobin depends on parasite genotype
Abstract
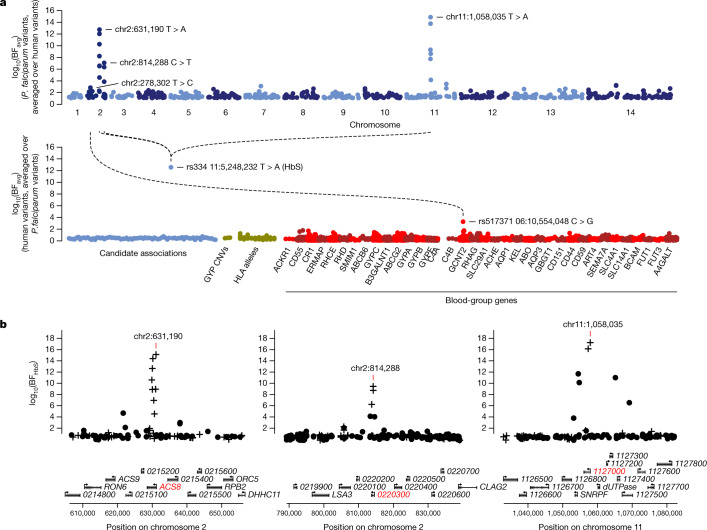
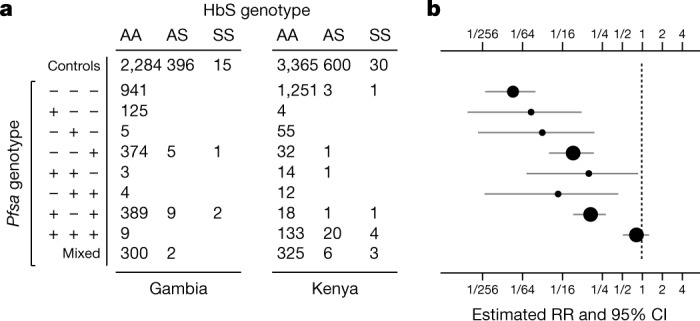
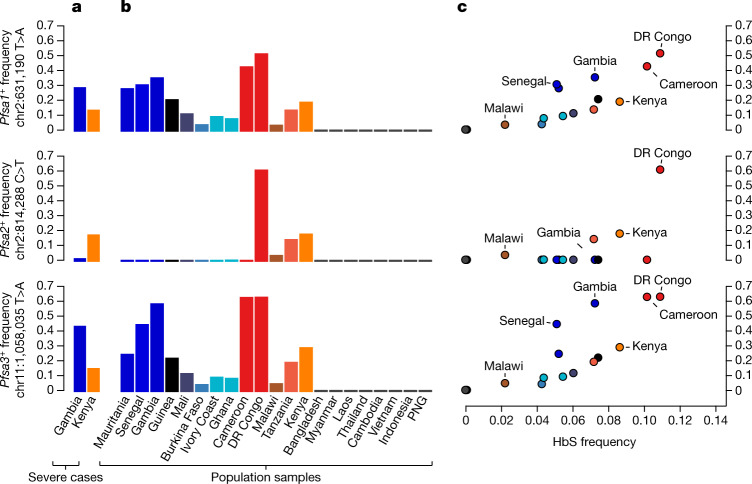
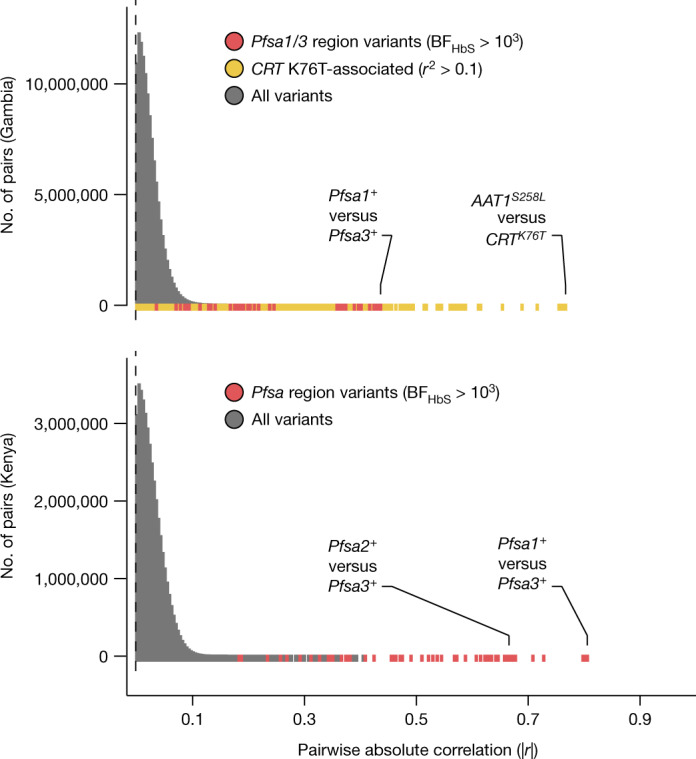
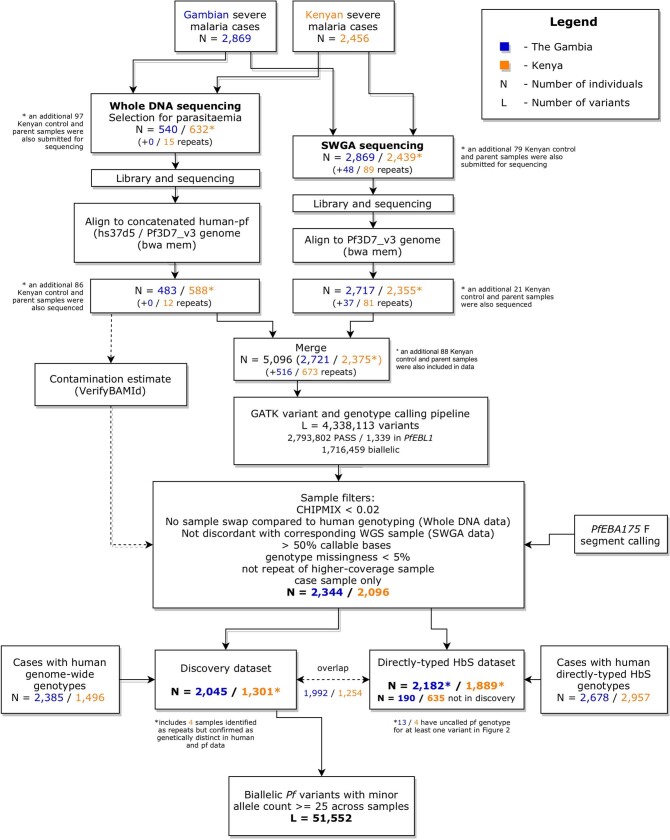
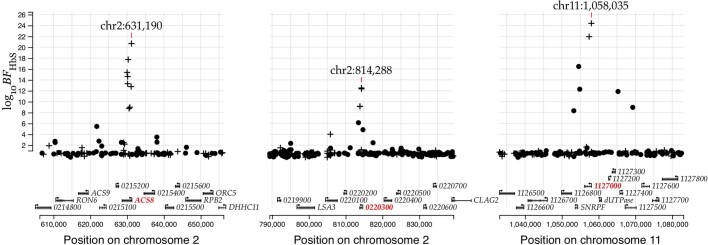
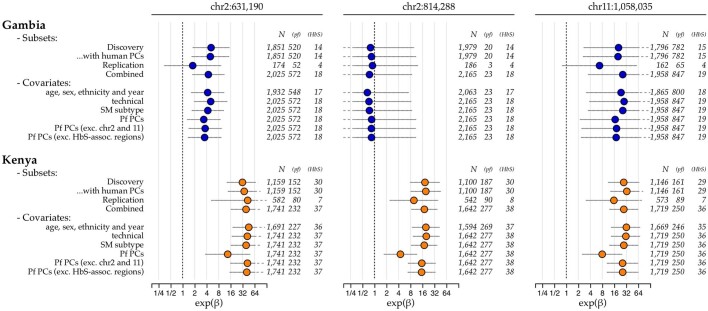
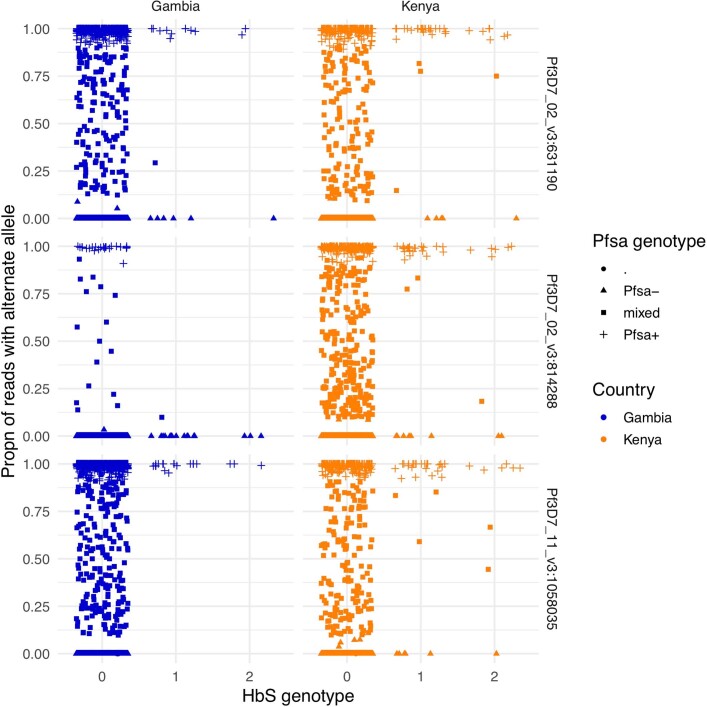
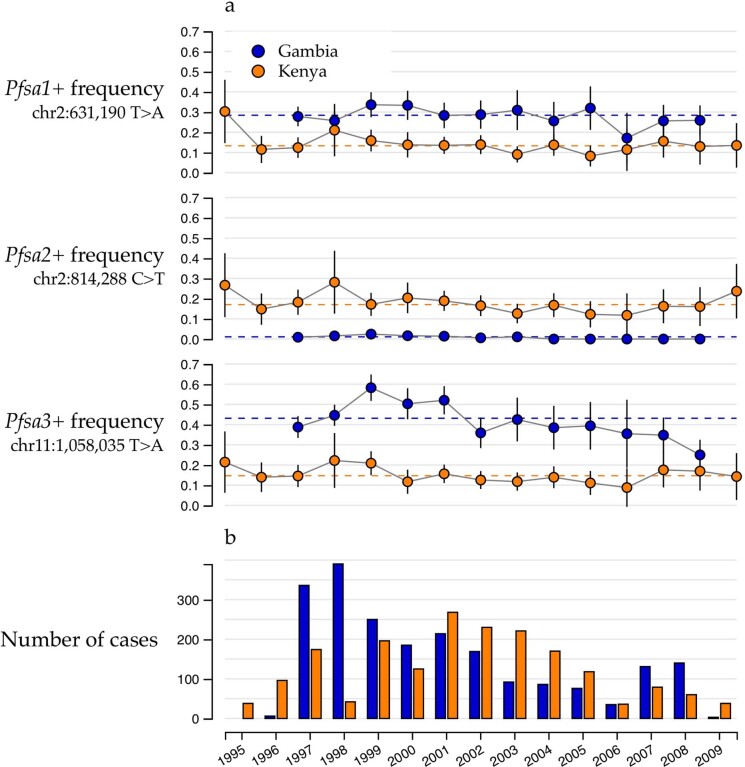
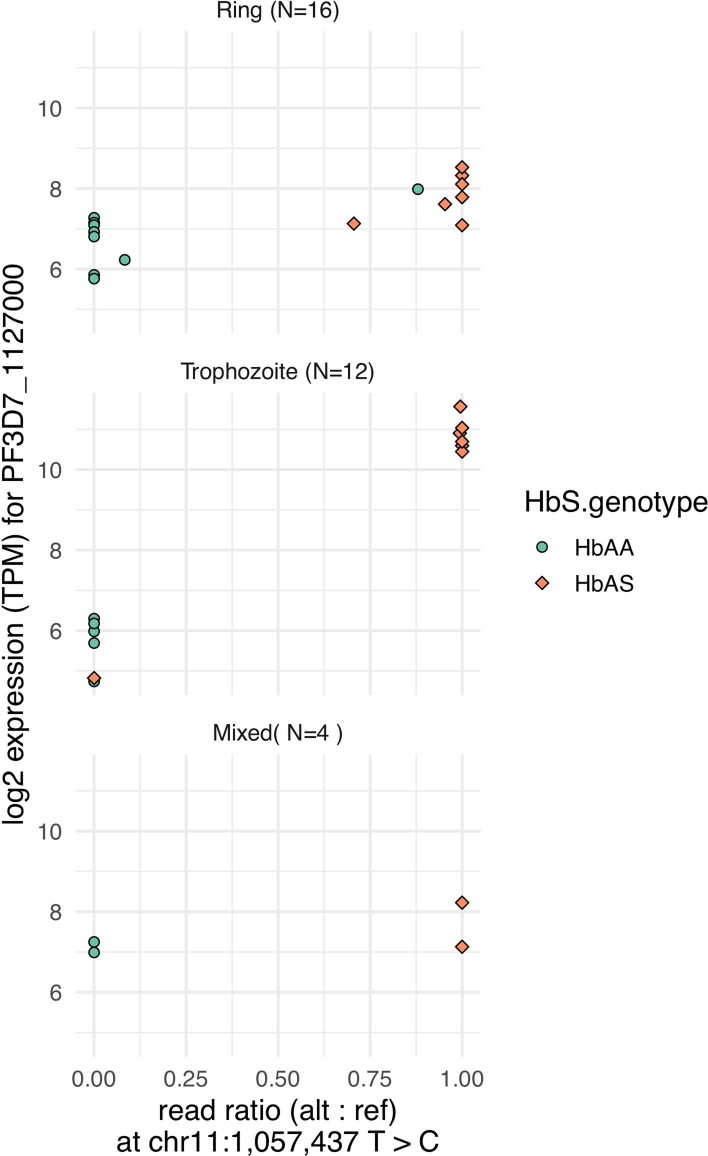
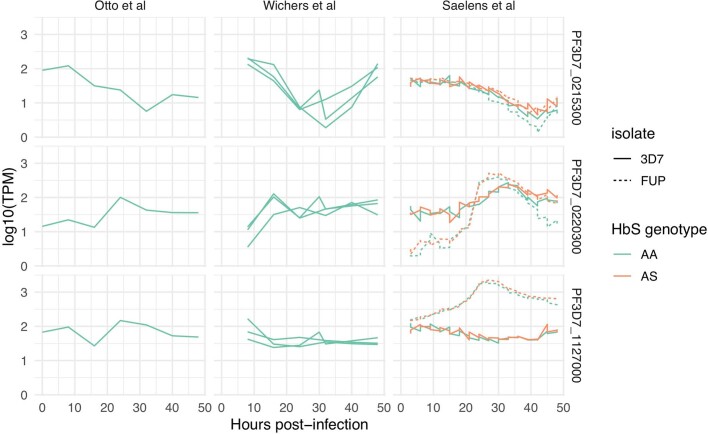
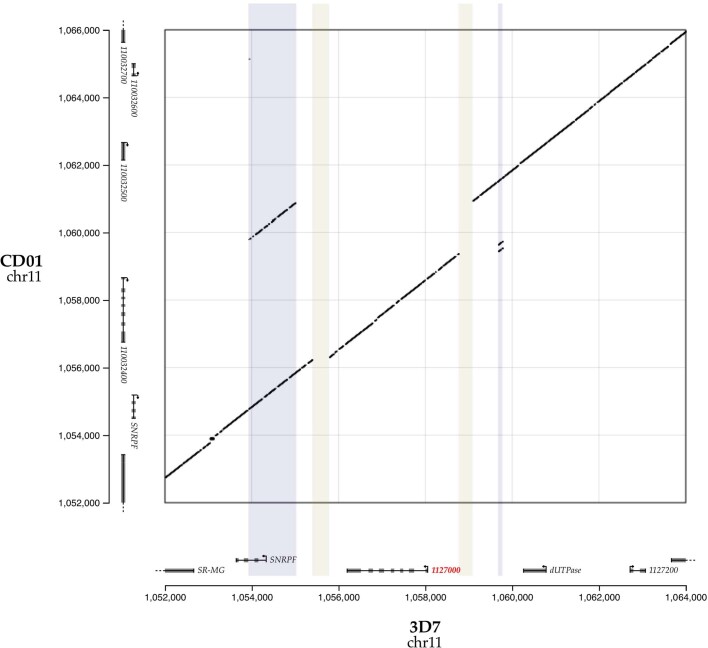
Host genetic factors can confer resistance against malaria1, raising the question of whether this has led to evolutionary adaptation of parasite populations. Here we searched for association between candidate host and parasite genetic variants in 3,346 Gambian and Kenyan children with severe malaria caused by Plasmodium falciparum. We identified a strong association between sickle haemoglobin (HbS) in the host and three regions of the parasite genome, which is not explained by population structure or other covariates, and which is replicated in additional samples. The HbS-associated alleles include nonsynonymous variants in the gene for the acyl-CoA synthetase family member2-4 PfACS8 on chromosome 2, in a second region of chromosome 2, and in a region containing structural variation on chromosome 11. The alleles are in strong linkage disequilibrium and have frequencies that covary with the frequency of HbS across populations, in particular being much more common in Africa than other parts of the world. The estimated protective effect of HbS against severe malaria, as determined by comparison of cases with population controls, varies greatly according to the parasite genotype at these three loci. These findings open up a new avenue of enquiry into the biological and epidemiological significance of the HbS-associated polymorphisms in the parasite genome and the evolutionary forces that have led to their high frequency and strong linkage disequilibrium in African P. falciparum populations.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Bethke LL, et al. Duplication, gene conversion, and genetic diversity in the species-specific acyl-CoA synthetase gene family of Plasmodium falciparum. Mol. Biochem. Parasitol. 2006;150:10–24. - PubMed
-
- Matesanz F, Téllez MA-D-M, Alcina A. The Plasmodium falciparum fatty acyl-CoA synthetase family (PfACS) and differential stage-specific expression in infected erythrocytes. Mol. Biochem. Parasitol. 2003;126:109–112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G0600230/MRC_/Medical Research Council/United Kingdom
- 202800/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- G0600718/MRC_/Medical Research Council/United Kingdom
- 090770/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- WT077383/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- UL1 TR002553/TR/NCATS NIH HHS/United States
- MC_UP_A900_1119/MRC_/Medical Research Council/United Kingdom
- 204911/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- R21 AI125988/AI/NIAID NIH HHS/United States
- MC_UP_A900_1118/MRC_/Medical Research Council/United Kingdom
- 206194/WT_/Wellcome Trust/United Kingdom
- 203141/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MR/M006212/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
