

Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity
- PMID: 34884612
- PMCID: PMC8657832
- DOI: 10.3390/ijms222312812
Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity
Abstract
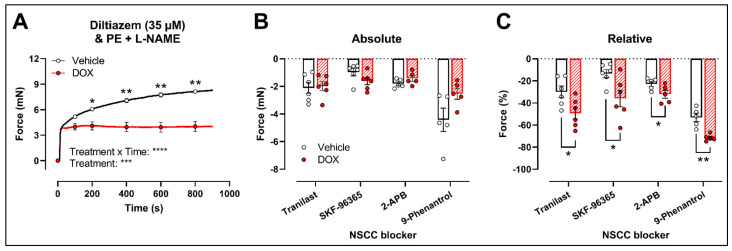
Clinical and animal studies have demonstrated that chemotherapeutic doxorubicin (DOX) increases arterial stiffness, a predictor of cardiovascular risk. Despite consensus about DOX-impaired endothelium-dependent vasodilation as a contributing mechanism, some studies have reported conflicting results on vascular smooth muscle cell (VSMC) function after DOX treatment. The present study aimed to investigate the effects of DOX on VSMC function. To this end, mice received a single injection of 4 mg DOX/kg, or mouse aortic segments were treated ex vivo with 1 μM DOX, followed by vascular reactivity evaluation 16 h later. Phenylephrine (PE)-induced VSMC contraction was decreased after DOX treatment. DOX did not affect the transient PE contraction dependent on Ca2+ release from the sarcoplasmic reticulum (0 mM Ca2+), but it reduced the subsequent tonic phase characterised by Ca2+ influx. These findings were supported by similar angiotensin II and attenuated endothelin-1 contractions. The involvement of voltage-gated Ca2+ channels in DOX-decreased contraction was excluded by using levcromakalim and diltiazem in PE-induced contraction and corroborated by similar K+ and serotonin contractions. Despite the evaluation of multiple blockers of transient receptor potential channels, the exact mechanism for DOX-decreased VSMC contraction remains elusive. Surprisingly, DOX reduced ex vivo but not in vivo arterial stiffness, highlighting the importance of appropriate timing for evaluating arterial stiffness in DOX-treated patients.
Keywords: arterial stiffness; cardio-oncology; doxorubicin; endothelial dysfunction; non-selective cation channel; vascular smooth muscle cell contraction.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures






Similar articles
-
Doxorubicin induces arterial stiffness: A comprehensive in vivo and ex vivo evaluation of vascular toxicity in mice.Toxicol Lett. 2021 Aug 1;346:23-33. doi: 10.1016/j.toxlet.2021.04.015. Epub 2021 Apr 22. Toxicol Lett. 2021. PMID: 33895255
-
Dexrazoxane does not mitigate early vascular toxicity induced by doxorubicin in mice.PLoS One. 2023 Nov 28;18(11):e0294848. doi: 10.1371/journal.pone.0294848. eCollection 2023. PLoS One. 2023. PMID: 38015959 Free PMC article.
-
Basal activity of voltage-gated Ca(2+) channels controls the IP3-mediated contraction by α(1)-adrenoceptor stimulation of mouse aorta segments.Eur J Pharmacol. 2015 Aug 5;760:163-71. doi: 10.1016/j.ejphar.2015.04.011. Epub 2015 Apr 22. Eur J Pharmacol. 2015. PMID: 25913240
-
Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels.J Cardiovasc Pharmacol. 2011 Feb;57(2):148-53. doi: 10.1097/FJC.0b013e3181f580d9. J Cardiovasc Pharmacol. 2011. PMID: 20729757 Review.
-
Calcium in Vascular Smooth Muscle Cell Elasticity and Adhesion: Novel Insights Into the Mechanism of Action.Front Physiol. 2019 Aug 7;10:852. doi: 10.3389/fphys.2019.00852. eCollection 2019. Front Physiol. 2019. PMID: 31440163 Free PMC article. Review.
Cited by
-
Doxorubicin-induced cardiovascular toxicity: a longitudinal evaluation of functional and molecular markers.Cardiovasc Res. 2023 Nov 25;119(15):2579-2590. doi: 10.1093/cvr/cvad136. Cardiovasc Res. 2023. PMID: 37625456 Free PMC article.
-
An integrative review of nonobvious puzzles of cellular and molecular cardiooncology.Cell Mol Biol Lett. 2023 May 23;28(1):44. doi: 10.1186/s11658-023-00451-y. Cell Mol Biol Lett. 2023. PMID: 37221467 Free PMC article. Review.
-
NADPH Oxidases in Cancer Therapy-Induced Cardiotoxicity: Mechanisms and Therapeutic Approaches.Cardiovasc Toxicol. 2025 Apr;25(4):631-649. doi: 10.1007/s12012-025-09976-4. Epub 2025 Feb 18. Cardiovasc Toxicol. 2025. PMID: 39966326 Review.
-
Doxorubicin-Induced Cardiac Remodeling: Mechanisms and Mitigation Strategies.Cardiovasc Drugs Ther. 2025 Feb 26. doi: 10.1007/s10557-025-07673-6. Online ahead of print. Cardiovasc Drugs Ther. 2025. PMID: 40009315 Review.
-
Elabela blunts doxorubicin-induced oxidative stress and ferroptosis in rat aortic adventitial fibroblasts by activating the KLF15/GPX4 signaling.Cell Stress Chaperones. 2023 Jan;28(1):91-103. doi: 10.1007/s12192-022-01317-6. Epub 2022 Dec 13. Cell Stress Chaperones. 2023. PMID: 36510036 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
