

The Role of EREG/EGFR Pathway in Tumor Progression
- PMID: 34884633
- PMCID: PMC8657471
- DOI: 10.3390/ijms222312828
The Role of EREG/EGFR Pathway in Tumor Progression
Abstract
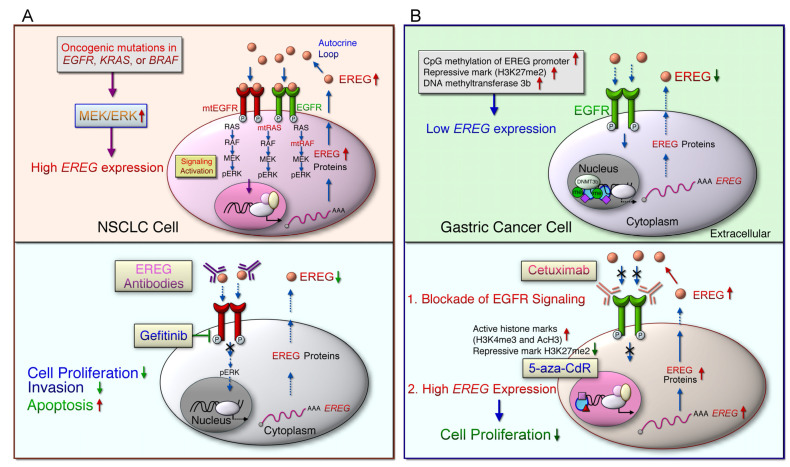
Aberrant activation of the epidermal growth factor receptor (EGFR/ERBB1) by erythroblastic leukemia viral oncogene homolog (ERBB) ligands contributes to various tumor malignancies, including lung cancer and colorectal cancer (CRC). Epiregulin (EREG) is one of the EGFR ligands and is low expressed in most normal tissues. Elevated EREG in various cancers mainly activates EGFR signaling pathways and promotes cancer progression. Notably, a higher EREG expression level in CRC with wild-type Kirsten rat sarcoma viral oncogene homolog (KRAS) is related to better efficacy of therapeutic treatment. By contrast, the resistance of anti-EGFR therapy in CRC was driven by low EREG expression, aberrant genetic mutation and signal pathway alterations. Additionally, EREG overexpression in non-small cell lung cancer (NSCLC) is anticipated to be a therapeutic target for EGFR-tyrosine kinase inhibitor (EGFR-TKI). However, recent findings indicate that EREG derived from macrophages promotes NSCLC cell resistance to EGFR-TKI treatment. The emerging events of EREG-mediated tumor promotion signals are generated by autocrine and paracrine loops that arise from tumor epithelial cells, fibroblasts, and macrophages in the tumor microenvironment (TME). The TME is a crucial element for the development of various cancer types and drug resistance. The regulation of EREG/EGFR pathways depends on distinct oncogenic driver mutations and cell contexts that allows specific pharmacological targeting alone or combinational treatment for tailored therapy. Novel strategies targeting EREG/EGFR, tumor-associated macrophages, and alternative activation oncoproteins are under development or undergoing clinical trials. In this review, we summarize the clinical outcomes of EREG expression and the interaction of this ligand in the TME. The EREG/EGFR pathway may be a potential target and may be combined with other driver mutation targets to combat specific cancers.
Keywords: cancer therapy; epidermal growth factor receptor (EGFR); epiregulin (EREG); tumor microenvironment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
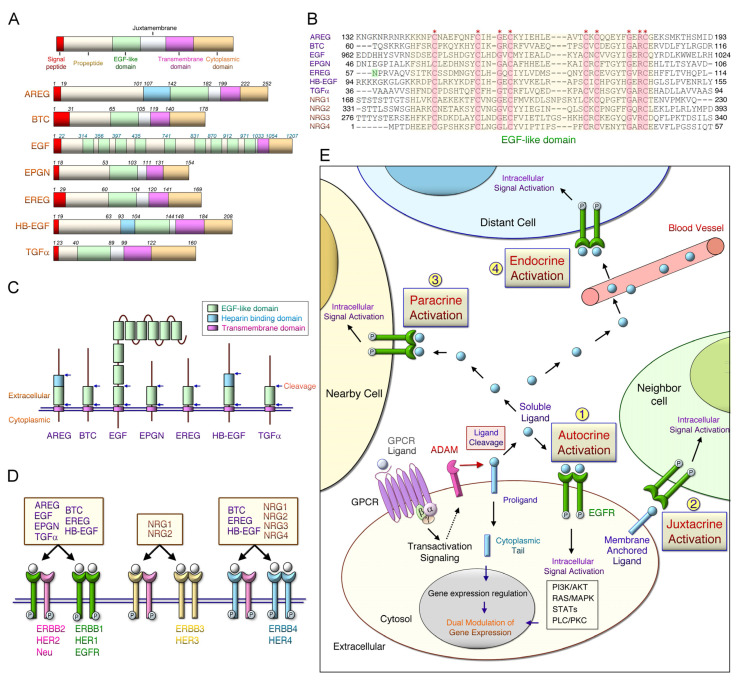
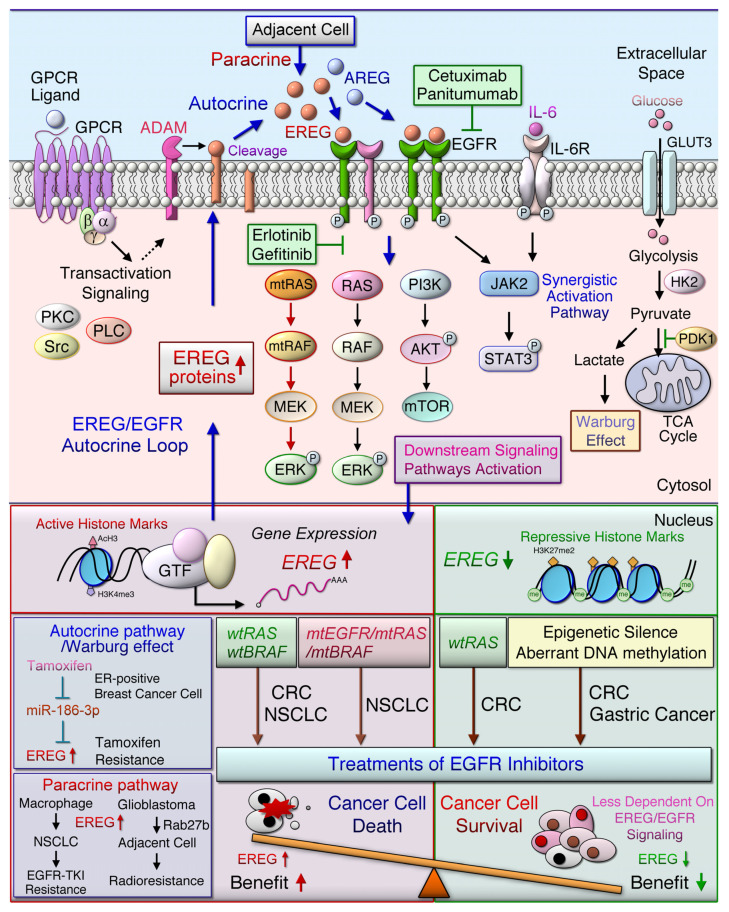
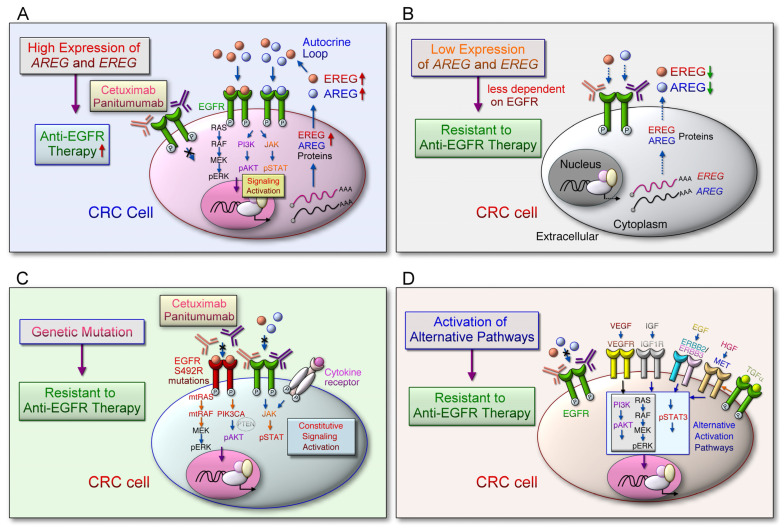
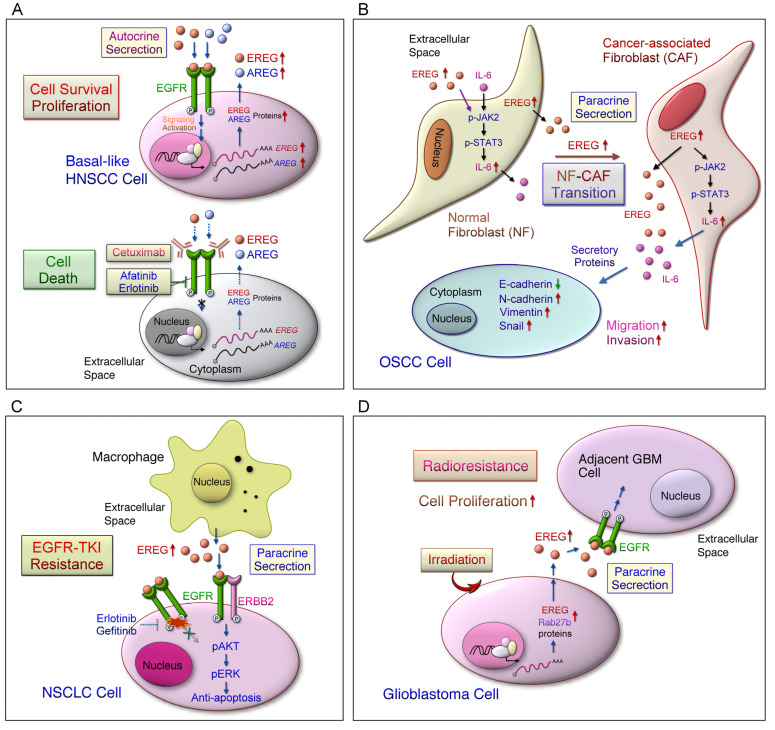
References
-
- Berdiel-Acer M., Maia A., Hristova Z., Borgoni S., Vetter M., Burmester S., Becki C., Michels B., Abnaof K., Binenbaum I., et al. Stromal NRG1 in luminal breast cancer defines pro-fibrotic and migratory cancer-associated fibroblasts. Oncogene. 2021;40:2651–2666. doi: 10.1038/s41388-021-01719-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
