

Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods
- PMID: 34884967
- PMCID: PMC8658603
- DOI: 10.3390/ijms222313162
Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods
Abstract
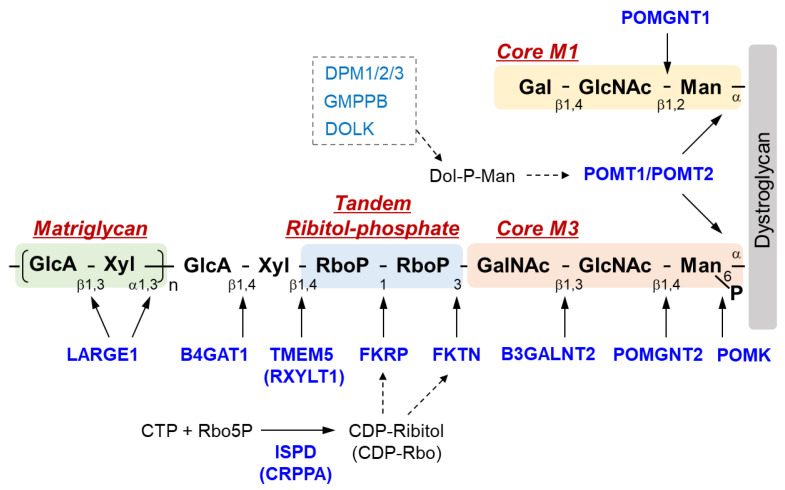
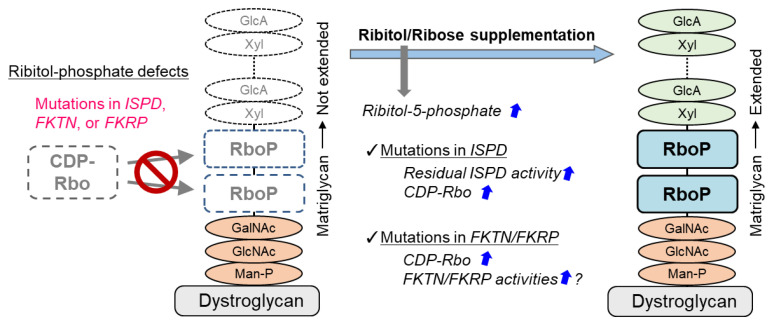
Dystroglycanopathy is a collective term referring to muscular dystrophies with abnormal glycosylation of dystroglycan. At least 18 causative genes of dystroglycanopathy have been identified, and its clinical symptoms are diverse, ranging from severe congenital to adult-onset limb-girdle types. Moreover, some cases are associated with symptoms involving the central nervous system. In the 2010s, the structure of sugar chains involved in the onset of dystroglycanopathy and the functions of its causative gene products began to be identified as if they were filling the missing pieces of a jigsaw puzzle. In parallel with these discoveries, various dystroglycanopathy model mice had been created, which led to the elucidation of its pathological mechanisms. Then, treatment strategies based on the molecular basis of glycosylation began to be proposed after the latter half of the 2010s. This review briefly explains the sugar chain structure of dystroglycan and the functions of the causative gene products of dystroglycanopathy, followed by introducing the pathological mechanisms involved as revealed from analyses of dystroglycanopathy model mice. Finally, potential therapeutic approaches based on the pathological mechanisms involved are discussed.
Keywords: dystroglycan; glycosylation; model mouse; muscular dystrophy; ribitol-phosphate; therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Muscular Dystrophy with Ribitol-Phosphate Deficiency: A Novel Post-Translational Mechanism in Dystroglycanopathy.J Neuromuscul Dis. 2017;4(4):259-267. doi: 10.3233/JND-170255. J Neuromuscul Dis. 2017. PMID: 29081423 Free PMC article. Review.
-
Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy.Hum Mol Genet. 2009 Feb 15;18(4):621-31. doi: 10.1093/hmg/ddn387. Epub 2008 Nov 18. Hum Mol Genet. 2009. PMID: 19017726 Free PMC article.
-
Mechanistic aspects of the formation of α-dystroglycan and therapeutic research for the treatment of α-dystroglycanopathy: A review.Mol Aspects Med. 2016 Oct;51:115-24. doi: 10.1016/j.mam.2016.07.003. Epub 2016 Jul 12. Mol Aspects Med. 2016. PMID: 27421908 Review.
-
Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies.Curr Opin Neurol. 2011 Oct;24(5):437-42. doi: 10.1097/WCO.0b013e32834a95e3. Curr Opin Neurol. 2011. PMID: 21825985 Review.
-
Temporal requirement of dystroglycan glycosylation during brain development and rescue of severe cortical dysplasia via gene delivery in the fetal stage.Hum Mol Genet. 2018 Apr 1;27(7):1174-1185. doi: 10.1093/hmg/ddy032. Hum Mol Genet. 2018. PMID: 29360985 Free PMC article.
Cited by
-
From adhesion complex to signaling hub: the dual role of dystroglycan.Front Mol Biosci. 2023 Dec 14;10:1325284. doi: 10.3389/fmolb.2023.1325284. eCollection 2023. Front Mol Biosci. 2023. PMID: 38155958 Free PMC article. Review.
-
Multi-parametric quantitative MRI of the lower limb muscles in a longitudinal study of limb-girdle muscular dystrophy R9.PLoS One. 2025 Apr 28;20(4):e0321463. doi: 10.1371/journal.pone.0321463. eCollection 2025. PLoS One. 2025. PMID: 40293996 Free PMC article.
-
GDP-Mannose Pyrophosphorylase B (GMPPB)-Related Disorders.Genes (Basel). 2023 Jan 31;14(2):372. doi: 10.3390/genes14020372. Genes (Basel). 2023. PMID: 36833299 Free PMC article. Review.
-
Impact of maternal compensation on developmental phenotypes in a zebrafish model of severe congenital muscular dystrophy.bioRxiv [Preprint]. 2025 May 13:2025.05.13.653769. doi: 10.1101/2025.05.13.653769. bioRxiv. 2025. PMID: 40463041 Free PMC article. Preprint.
-
CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy.Cells. 2023 Oct 12;12(20):2444. doi: 10.3390/cells12202444. Cells. 2023. PMID: 37887288 Free PMC article.
References
-
- Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E., et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2001;69:1198–1209. doi: 10.1086/324412. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
