

Sphingosine 1-Phosphate Receptor 4 Promotes Nonalcoholic Steatohepatitis by Activating NLRP3 Inflammasome
- PMID: 34890841
- PMCID: PMC8810559
- DOI: 10.1016/j.jcmgh.2021.12.002
Sphingosine 1-Phosphate Receptor 4 Promotes Nonalcoholic Steatohepatitis by Activating NLRP3 Inflammasome
Abstract
Background & aims: Sphingosine 1-phosphate receptors (S1PRs) are a group of G-protein-coupled receptors that confer a broad range of functional effects in chronic inflammatory and metabolic diseases. S1PRs also may mediate the development of nonalcoholic steatohepatitis (NASH), but the specific subtypes involved and the mechanism of action are unclear.
Methods: We investigated which type of S1PR isoforms is activated in various murine models of NASH. The mechanism of action of S1PR4 was examined in hepatic macrophages isolated from high-fat, high-cholesterol diet (HFHCD)-fed mice. We developed a selective S1PR4 functional antagonist by screening the fingolimod (2-amino-2-[2-(4- n -octylphenyl)ethyl]-1,3- propanediol hydrochloride)-like sphingolipid-focused library.
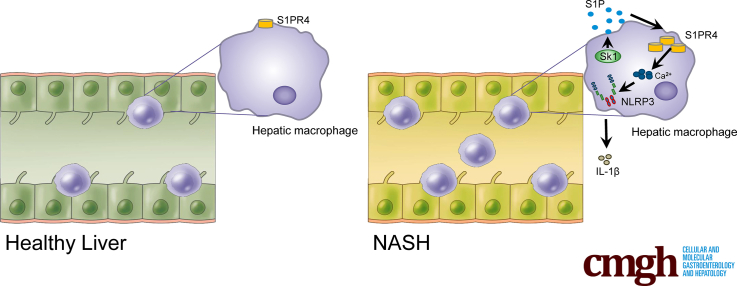
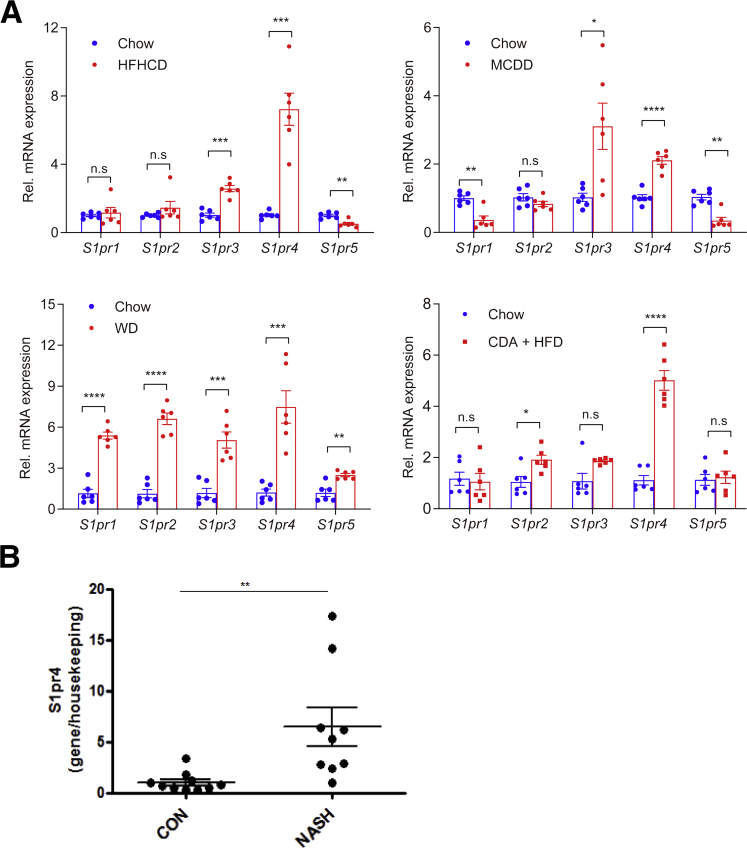
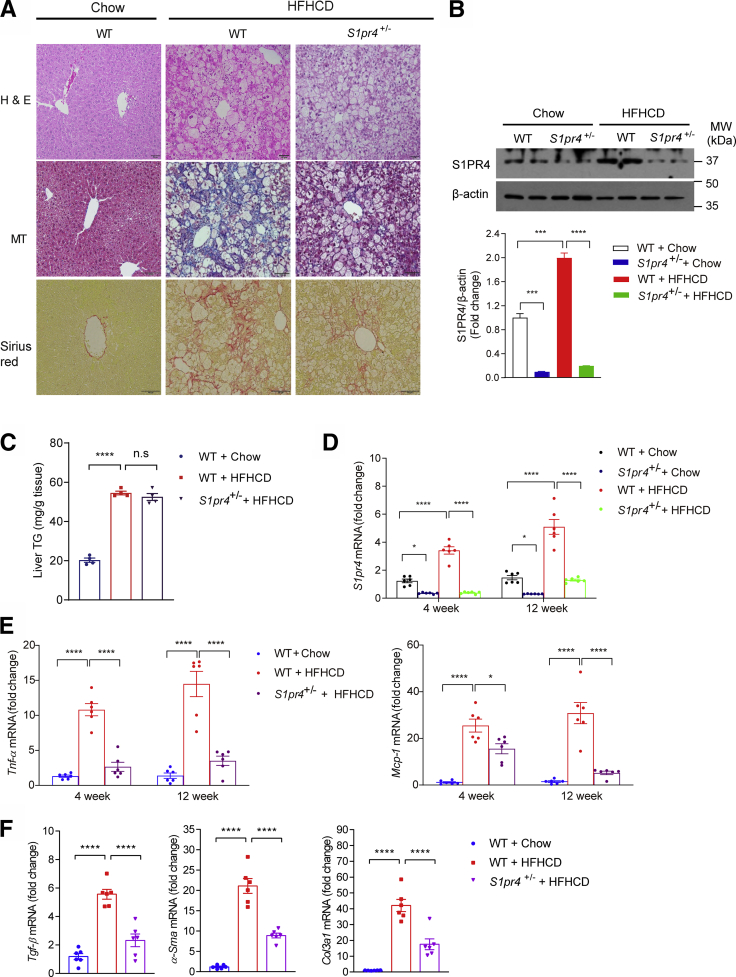
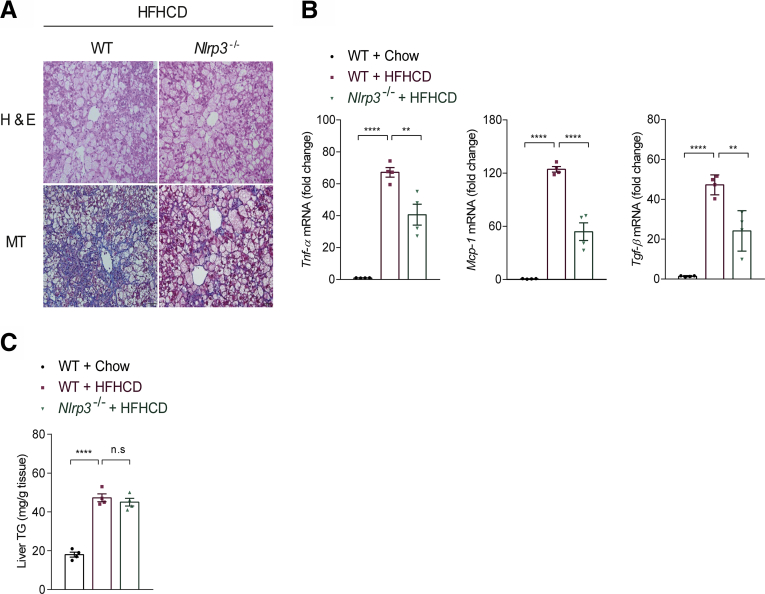
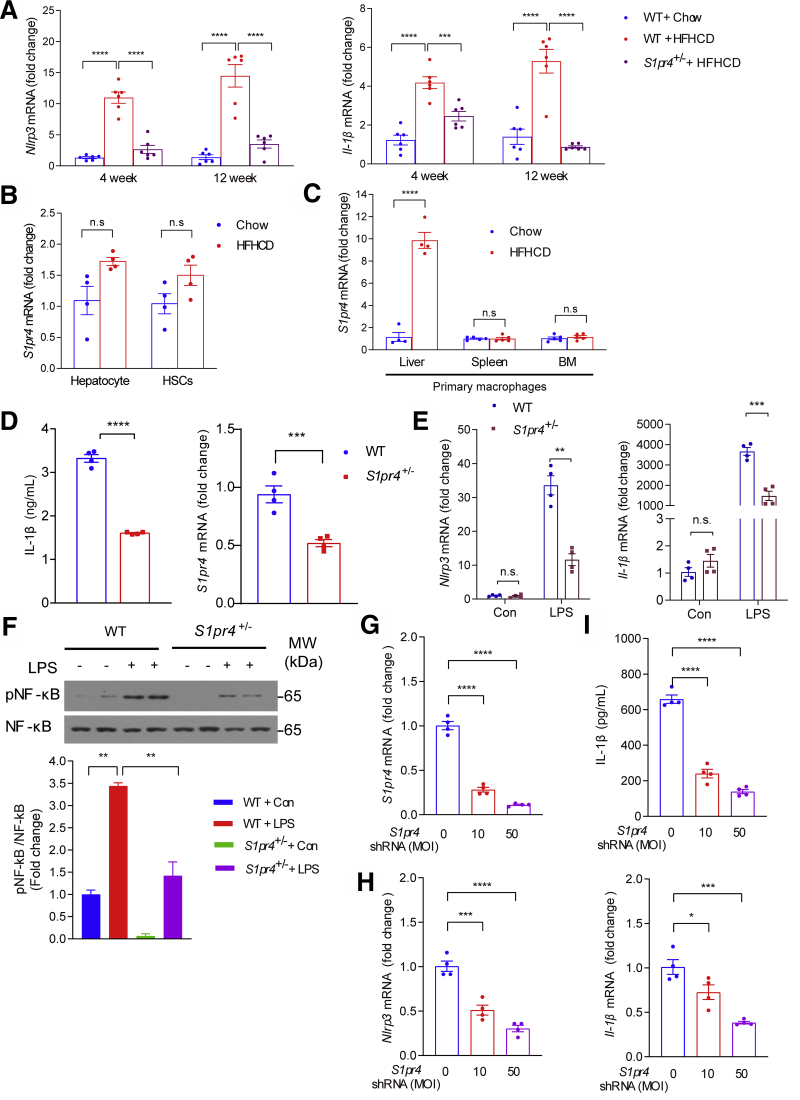
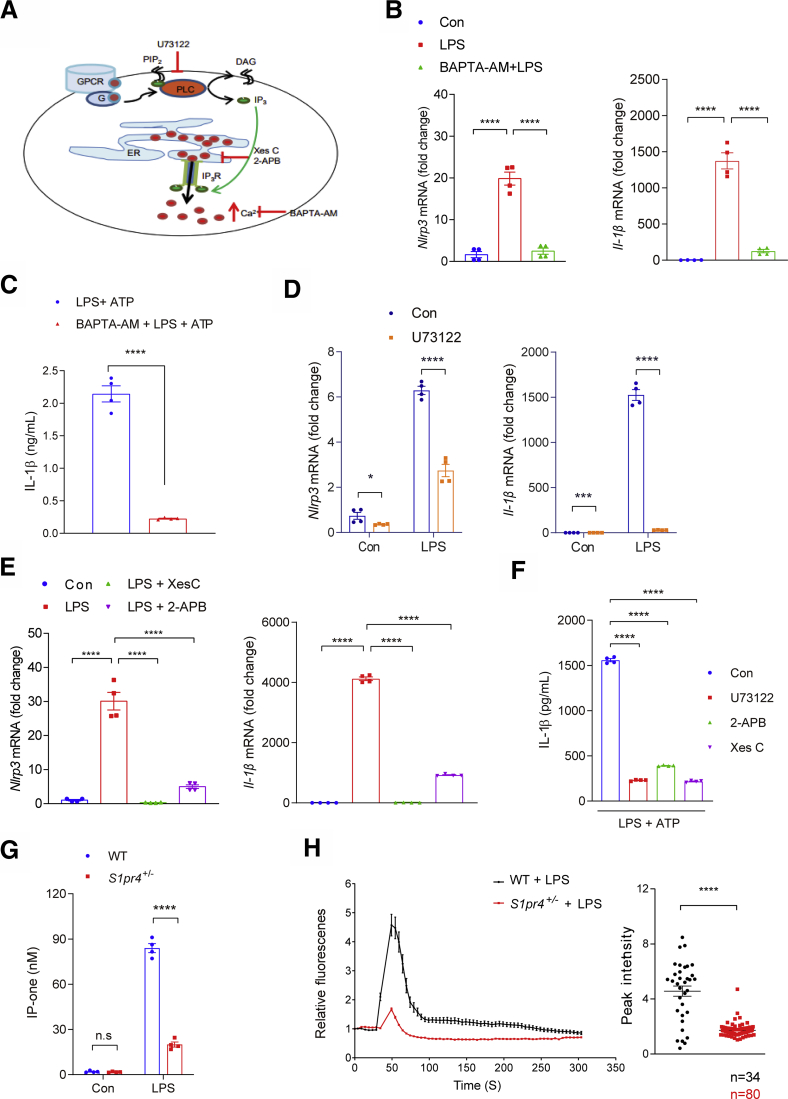
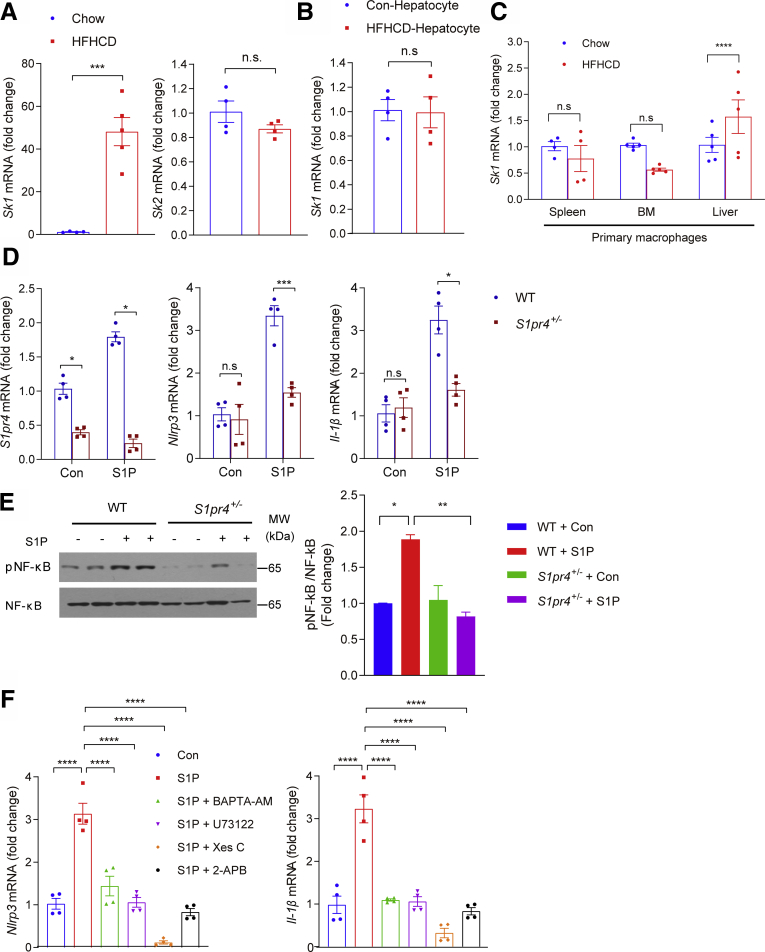
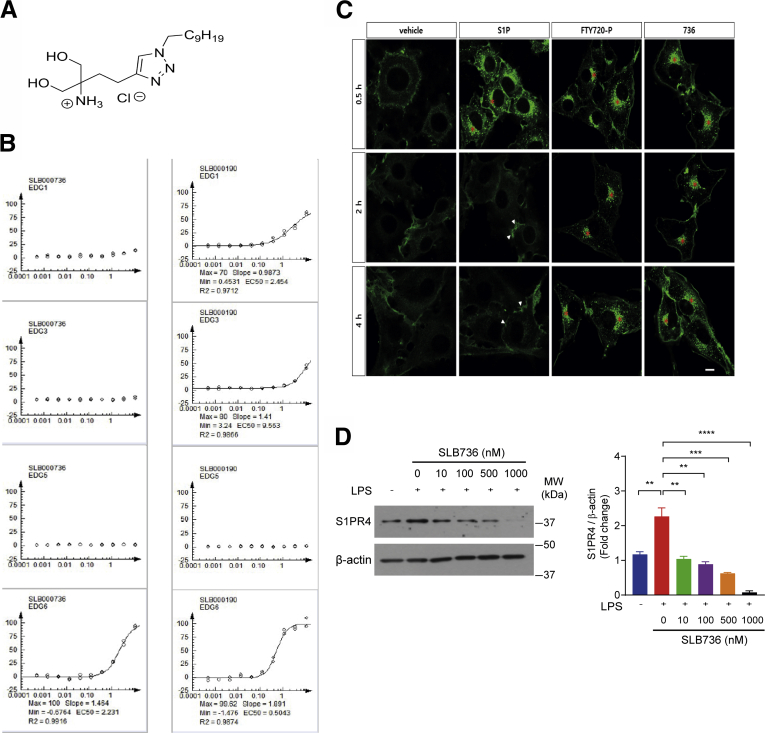
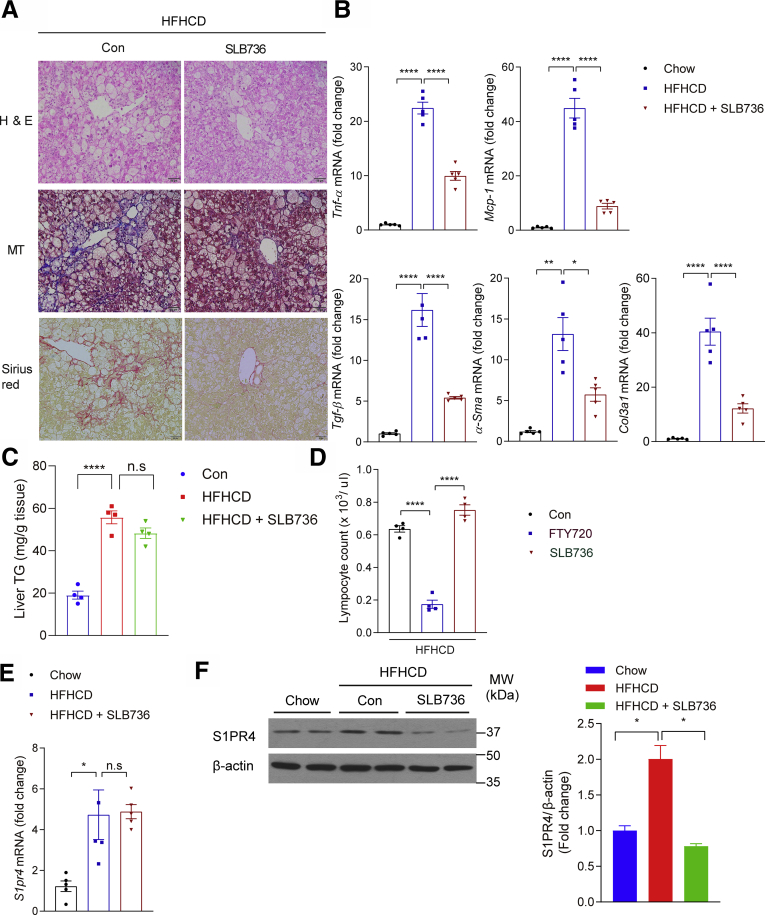
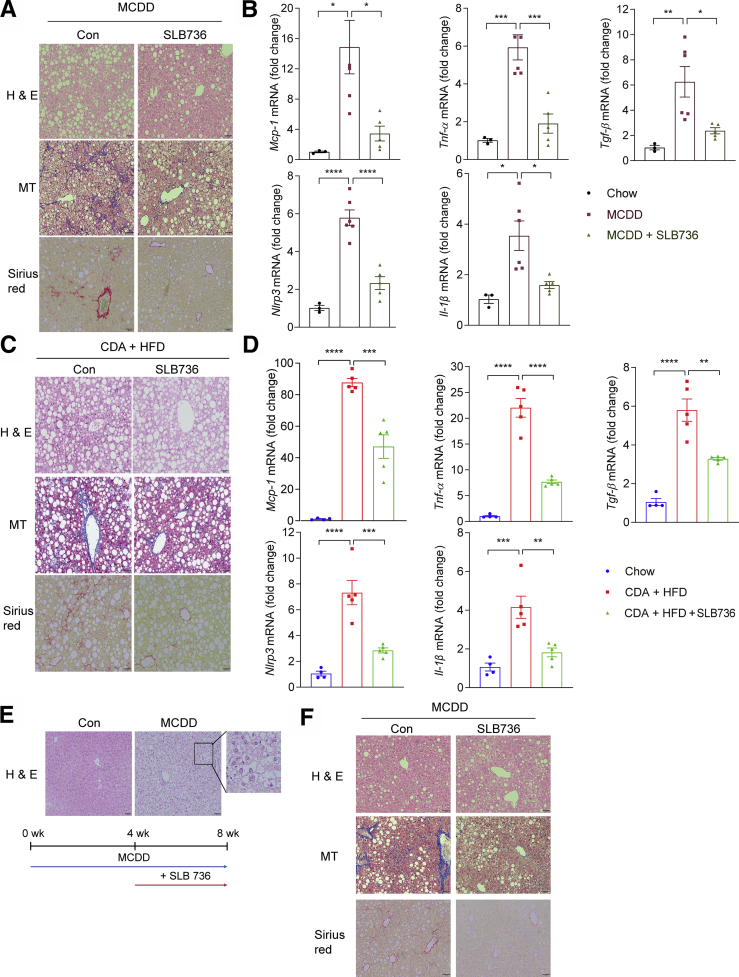
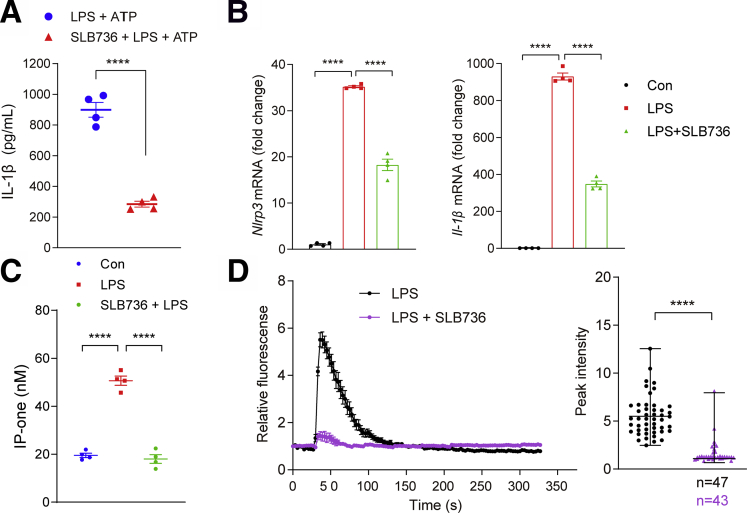
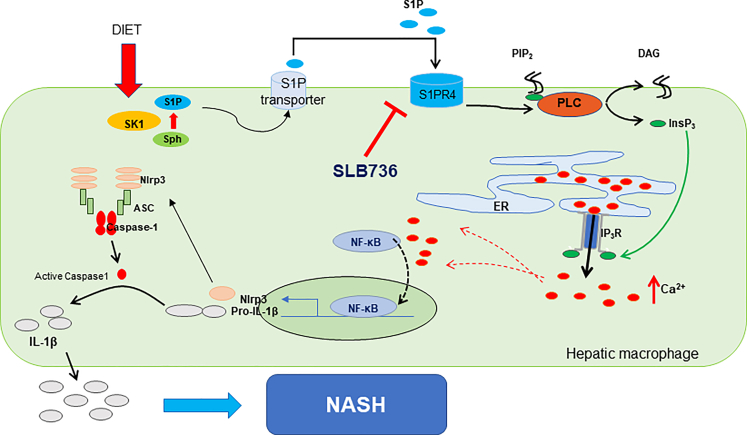
Results: The livers of various mouse models of NASH as well as hepatic macrophages showed high expression of S1pr4. Moreover, in a cohort of NASH patients, expression of S1PR4 was 6-fold higher than those of healthy controls. S1pr4+/- mice were protected from HFHCD-induced NASH and hepatic fibrosis without changes in steatosis. S1pr4 depletion in hepatic macrophages inhibited lipopolysaccharide-mediated Ca++ release and deactivated the Nod-like receptor pyrin domain-containning protein 3 (NLRP3) inflammasome. S1P increased the expression of S1pr4 in hepatic macrophages and activated NLRP3 inflammasome through inositol trisphosphate/inositol trisphosphate-receptor-dependent [Ca++] signaling. To further clarify the biological function of S1PR4, we developed SLB736, a novel selective functional antagonist of SIPR4. Similar to S1pr4+/- mice, administration of SLB736 to HFHCD-fed mice prevented the development of NASH and hepatic fibrosis, but not steatosis, by deactivating the NLRP3 inflammasome.
Conclusions: S1PR4 may be a new therapeutic target for NASH that mediates the activation of NLRP3 inflammasome in hepatic macrophages.
Keywords: Ca(++); Functional Antagonist; Hepatic Macrophages; S1P.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Lassailly G., Caiazzo R., Pattou F., Mathurin P. Perspectives on treatment for nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1835–1848. - PubMed
-
- Younossi Z.M., Loomba R., Rinella M.E., Bugianesi E., Marchesini G., Neuschwander-Tetri B.A., Serfaty L., Negro F., Caldwell S.H., Ratziu V., Corey K.E., Friedman S.L., Abdelmalek M.F., Harrison S.A., Sanyal A.J., Lavine J.E., Mathurin P., Charlton M.R., Chalasani N.P., Anstee Q.M., Kowdley K.V., George J., Goodman Z.D., Lindor K. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2018;68:361–371. - PMC - PubMed
-
- Koh E.H., Yoon J.E., Ko M.S., Leem J., Yun J.Y., Hong C.H., Cho Y.K., Lee S.E., Jang J.E., Baek J.Y., Yoo H.J., Kim S.J., Sung C.O., Lim J.S., Jeong W.I., Back S.H., Baek I.J., Torres S., Solsona-Vilarrasa E., Conde de la Rosa L., Garcia-Ruiz C., Feldstein A.E., Fernandez-Checa J.C., Lee K.U. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut. 2021;70:1954–1964. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
