

Pathogenic insights from genetic causes of autoinflammatory inflammasomopathies and interferonopathies
- PMID: 34893352
- PMCID: PMC8901451
- DOI: 10.1016/j.jaci.2021.10.027
Pathogenic insights from genetic causes of autoinflammatory inflammasomopathies and interferonopathies
Abstract
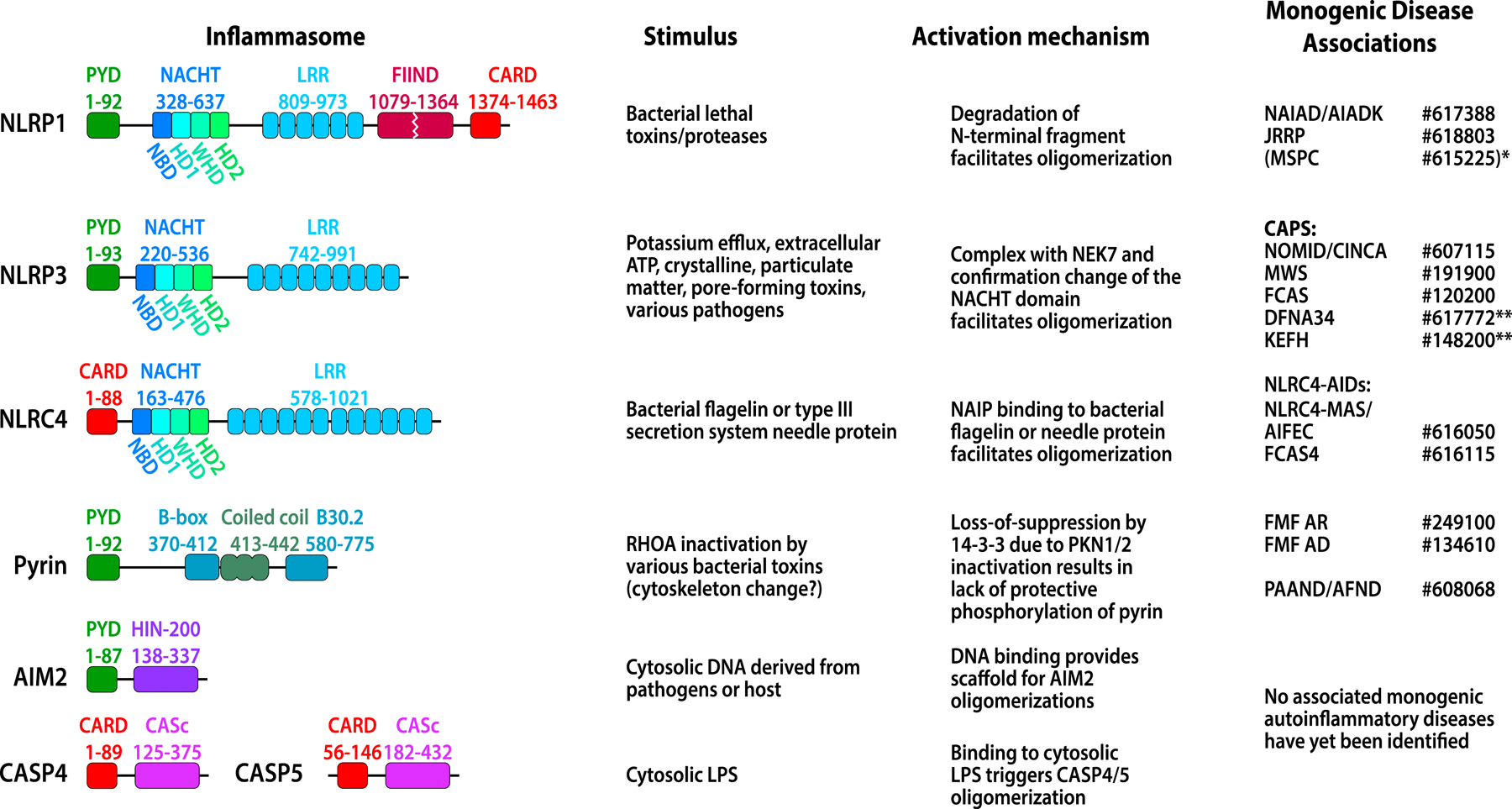
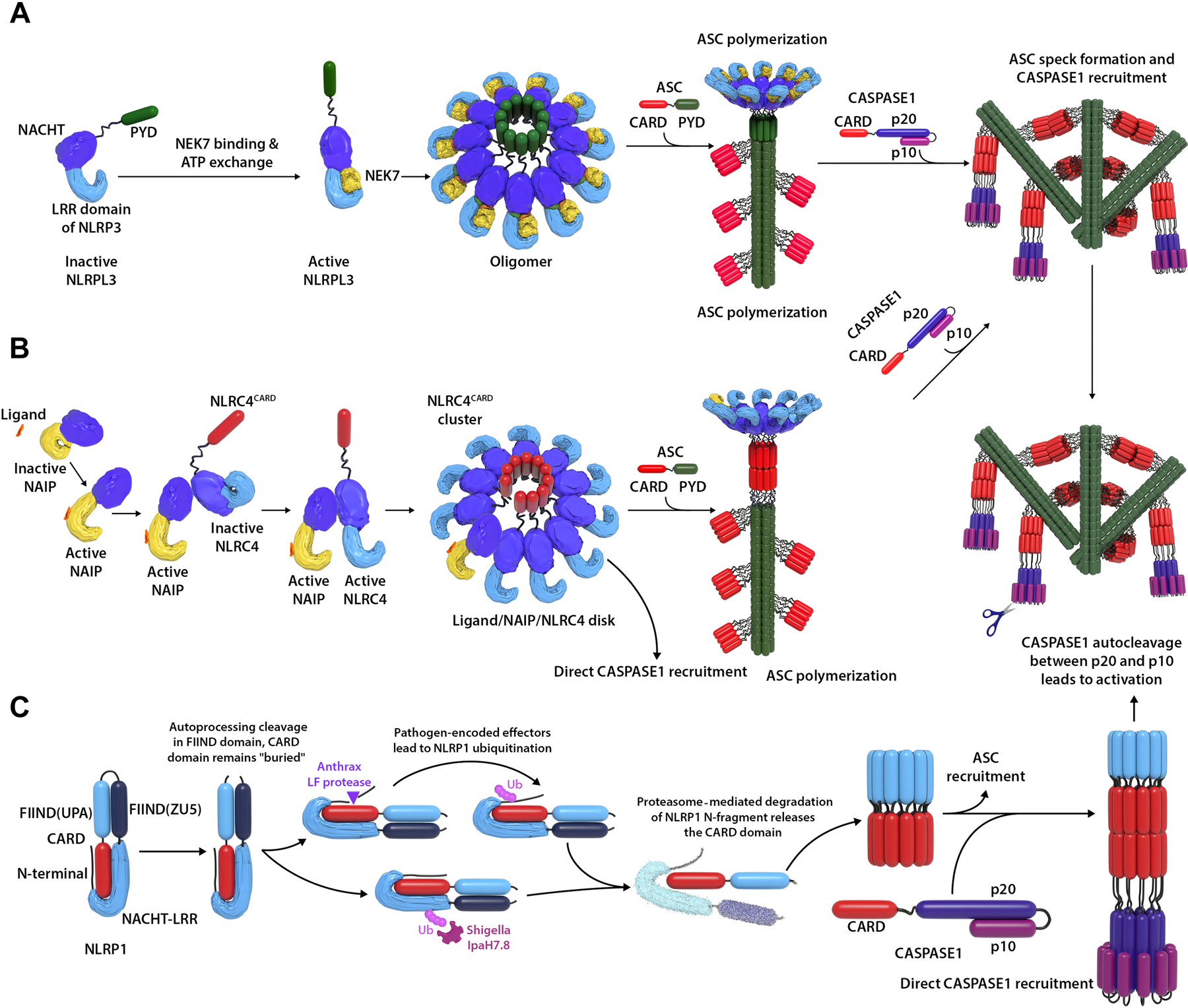
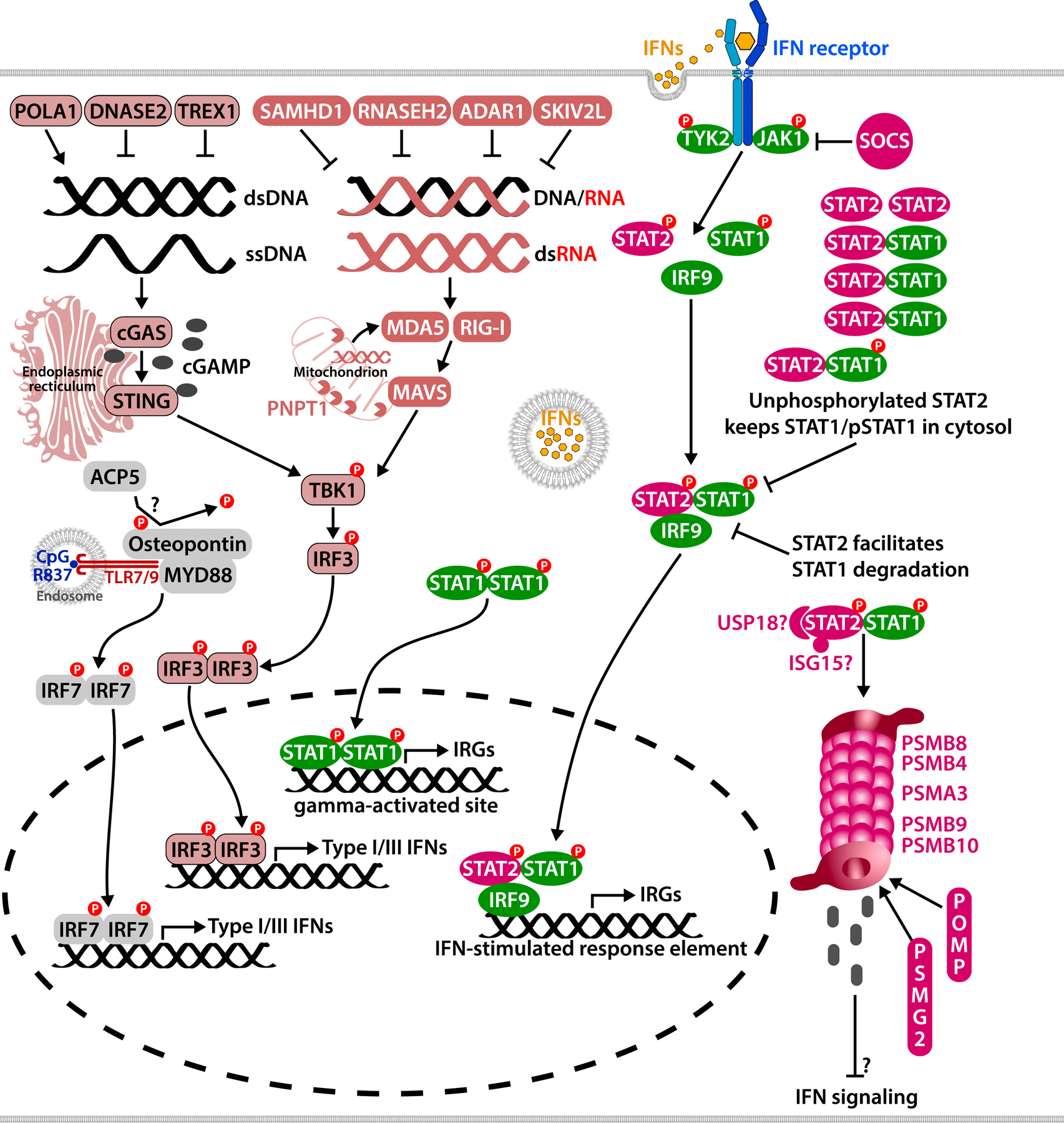
A number of systemic autoinflammatory diseases arise from gain-of-function mutations in genes encoding IL-1-activating inflammasomes or cytoplasmic nucleic acid sensors including the receptor and sensor STING and result in increased IL-1 and type I interferon production, respectively. Blocking these pathways in human diseases has provided proof-of-concept, confirming the prominent roles of these cytokines in disease pathogenesis. Recent insights into the multilayered regulation of these sensor pathways and insights into their role in amplifying the disease pathogenesis of monogenic and complex genetic diseases spurred new drug development targeting the sensors. This review provides insights into the pathogenesis and genetic causes of these "prototypic" diseases caused by gain-of function mutations in IL-1-activating inflammasomes (inflammasomopathies) and in interferon-activating pathways (interferonopathies) including STING-associated vasculopathy with onset in infancy, Aicardi-Goutieres syndrome, and proteasome-associated autoinflammatory syndromes that link activation of the viral sensors STING, "self" nucleic acid metabolism, and the ubiquitin-proteasome system to "type I interferon production" and human diseases. Clinical responses and biomarker changes to Janus kinase inhibitors confirm a role of interferons, and a growing number of diseases with "interferon signatures" unveil extensive cross-talk between major inflammatory pathways. Understanding these interactions promises new tools in tackling the significant clinical challenges in treating patients with these conditions.
Keywords: Aicardi-Goutieres syndrome; Interferonopathies; JAK inhibitor; STING; inflammasome; inflammasomopathies; interferon; proteasome-associated autoinflammatory syndrome.
Published by Elsevier Inc.
Conflict of interest statement
Conflict of interest:
Dr. Goldbach-Mansky has received study support under government CRADAs from SOBI, Regeneron, Novartis and Eli Lilly. All other authors have nothing to disclose.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
