

Bayesian Inference of Clonal Expansions in a Dated Phylogeny
- PMID: 34893904
- PMCID: PMC9366454
- DOI: 10.1093/sysbio/syab095
Bayesian Inference of Clonal Expansions in a Dated Phylogeny
Abstract
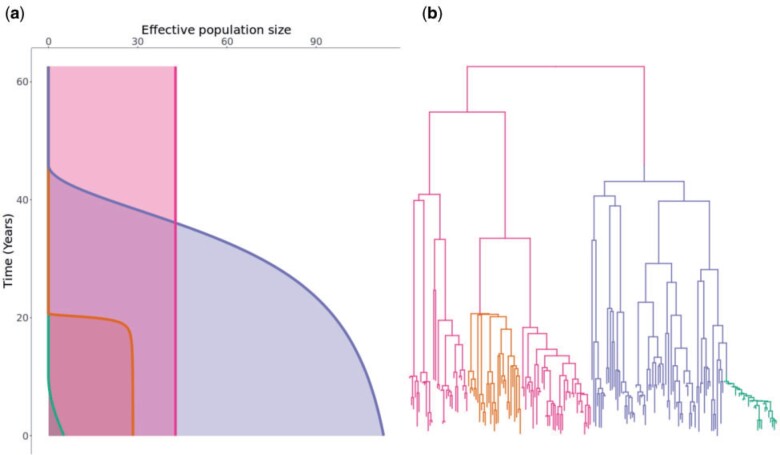
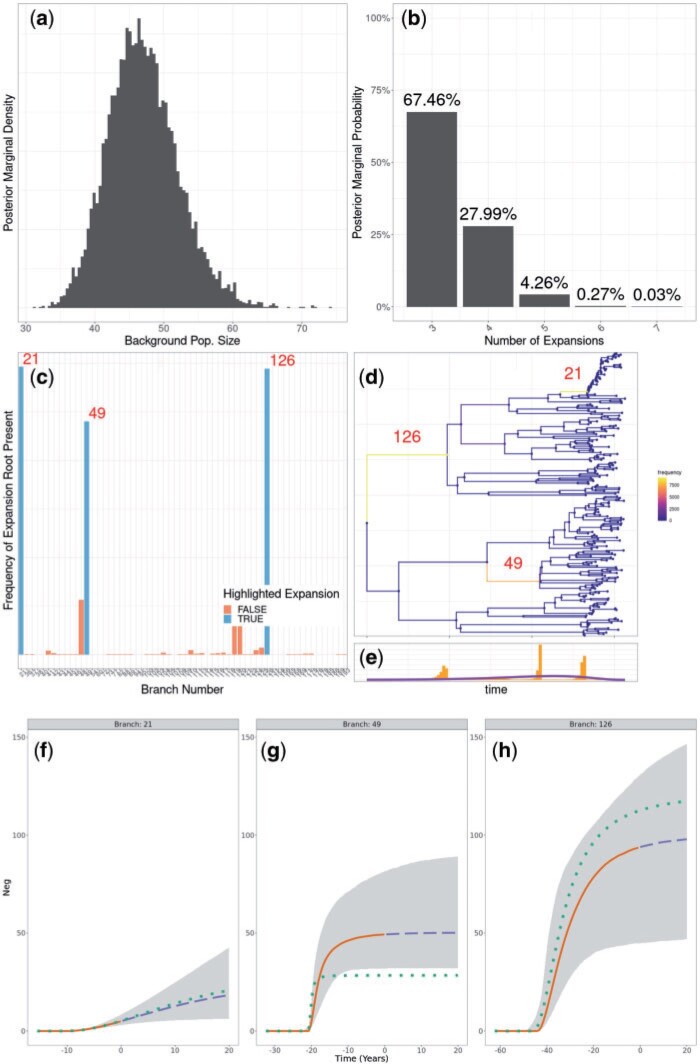
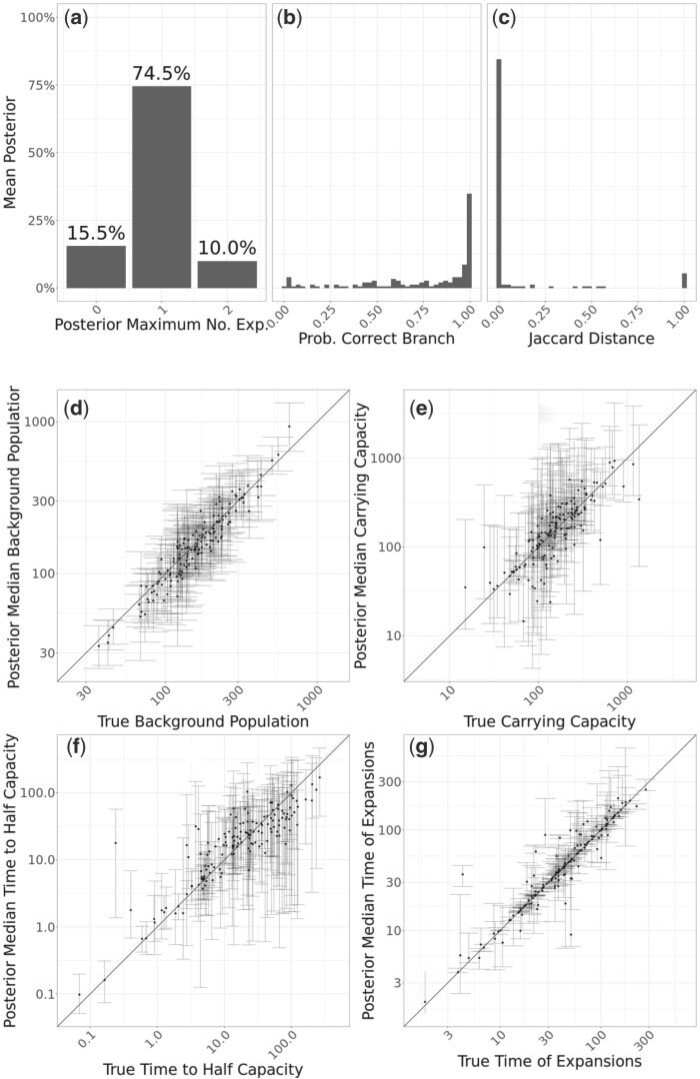
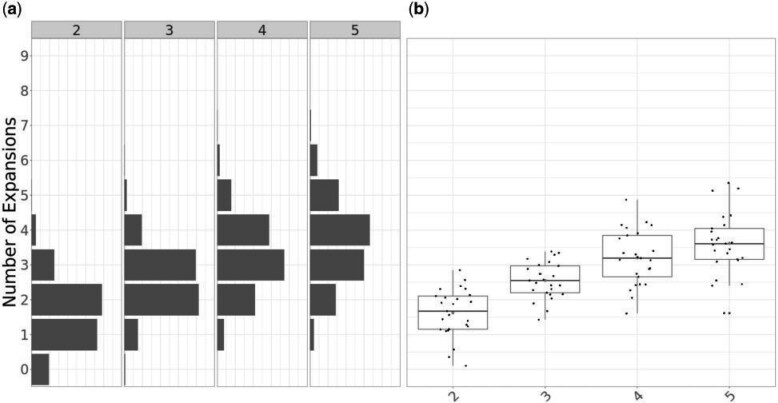
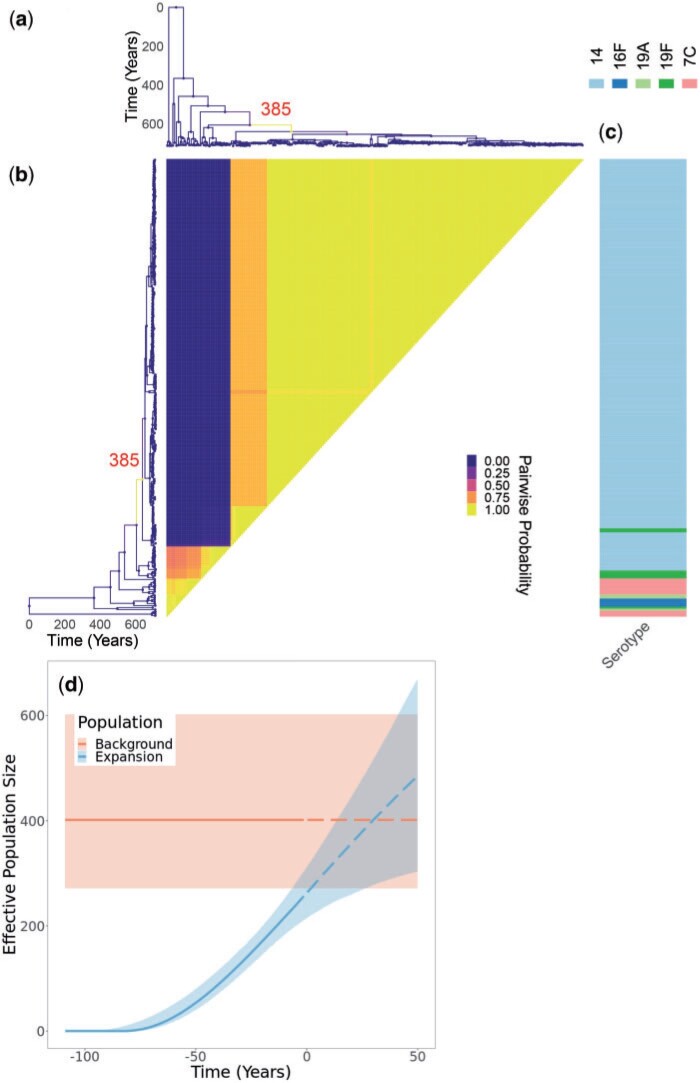
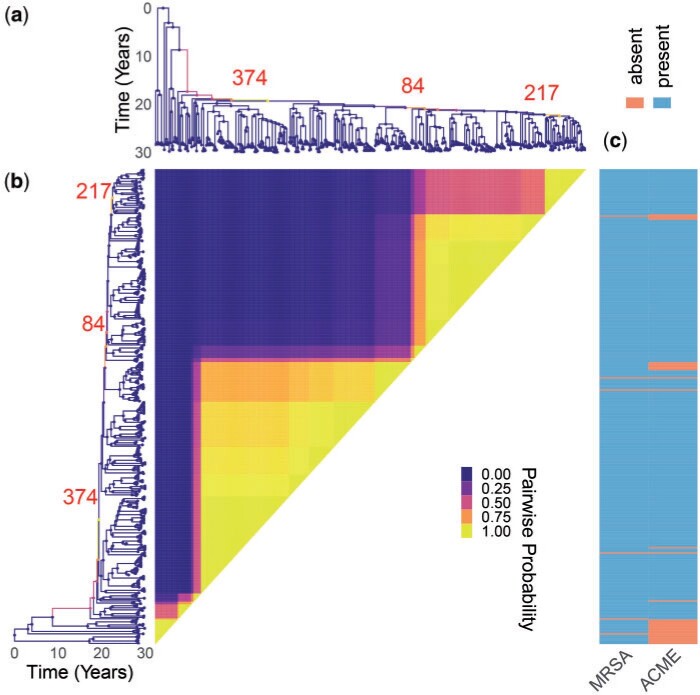
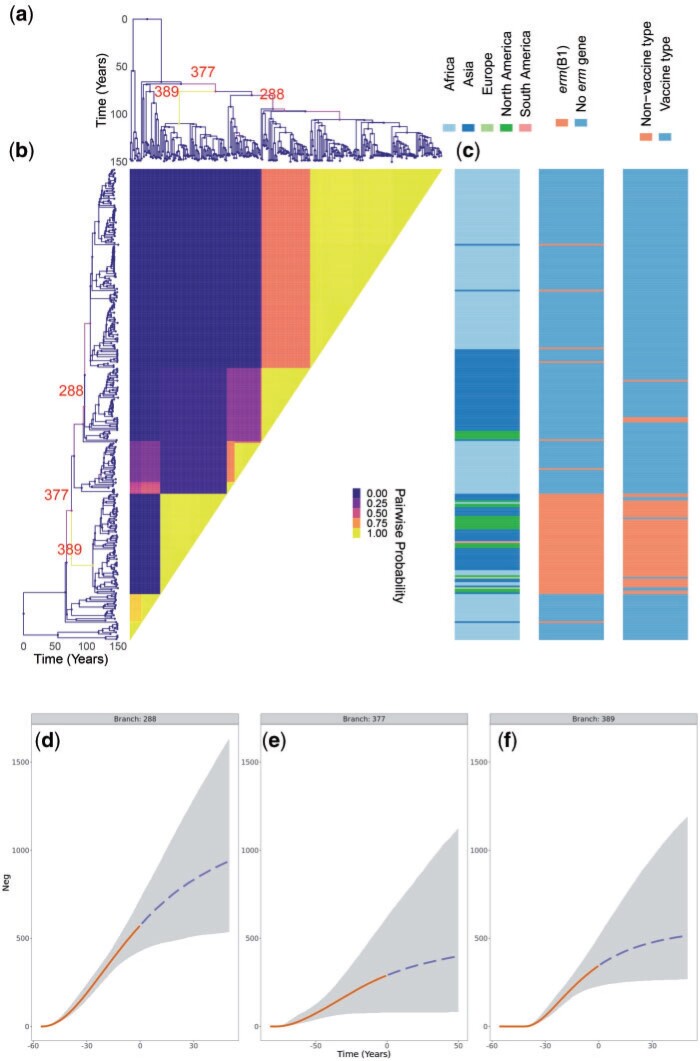
Microbial population genetics models often assume that all lineages are constrained by the same population size dynamics over time. However, many neutral and selective events can invalidate this assumption and can contribute to the clonal expansion of a specific lineage relative to the rest of the population. Such differential phylodynamic properties between lineages result in asymmetries and imbalances in phylogenetic trees that are sometimes described informally but which are difficult to analyze formally. To this end, we developed a model of how clonal expansions occur and affect the branching patterns of a phylogeny. We show how the parameters of this model can be inferred from a given dated phylogeny using Bayesian statistics, which allows us to assess the probability that one or more clonal expansion events occurred. For each putative clonal expansion event, we estimate its date of emergence and subsequent phylodynamic trajectory, including its long-term evolutionary potential which is important to determine how much effort should be placed on specific control measures. We demonstrate the applicability of our methodology on simulated and real data sets. Inference under our clonal expansion model can reveal important features in the evolution and epidemiology of infectious disease pathogens. [Clonal expansion; genomic epidemiology; microbial population genomics; phylodynamics.].
© The Author(s) 2021. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures
References
-
- Allen L. 2008. An introduction to stochastic epidemic models. In: Brauer F., editor. Mathematical epidemiology vol. 1945 of Lecture Notes in Mathematics. Berlin Heidelberg: Springer. p. 81–130.
