

The evolution of regulated cell death pathways in animals and their evasion by pathogens
- PMID: 34898294
- PMCID: PMC8676434
- DOI: 10.1152/physrev.00002.2021
The evolution of regulated cell death pathways in animals and their evasion by pathogens
Abstract
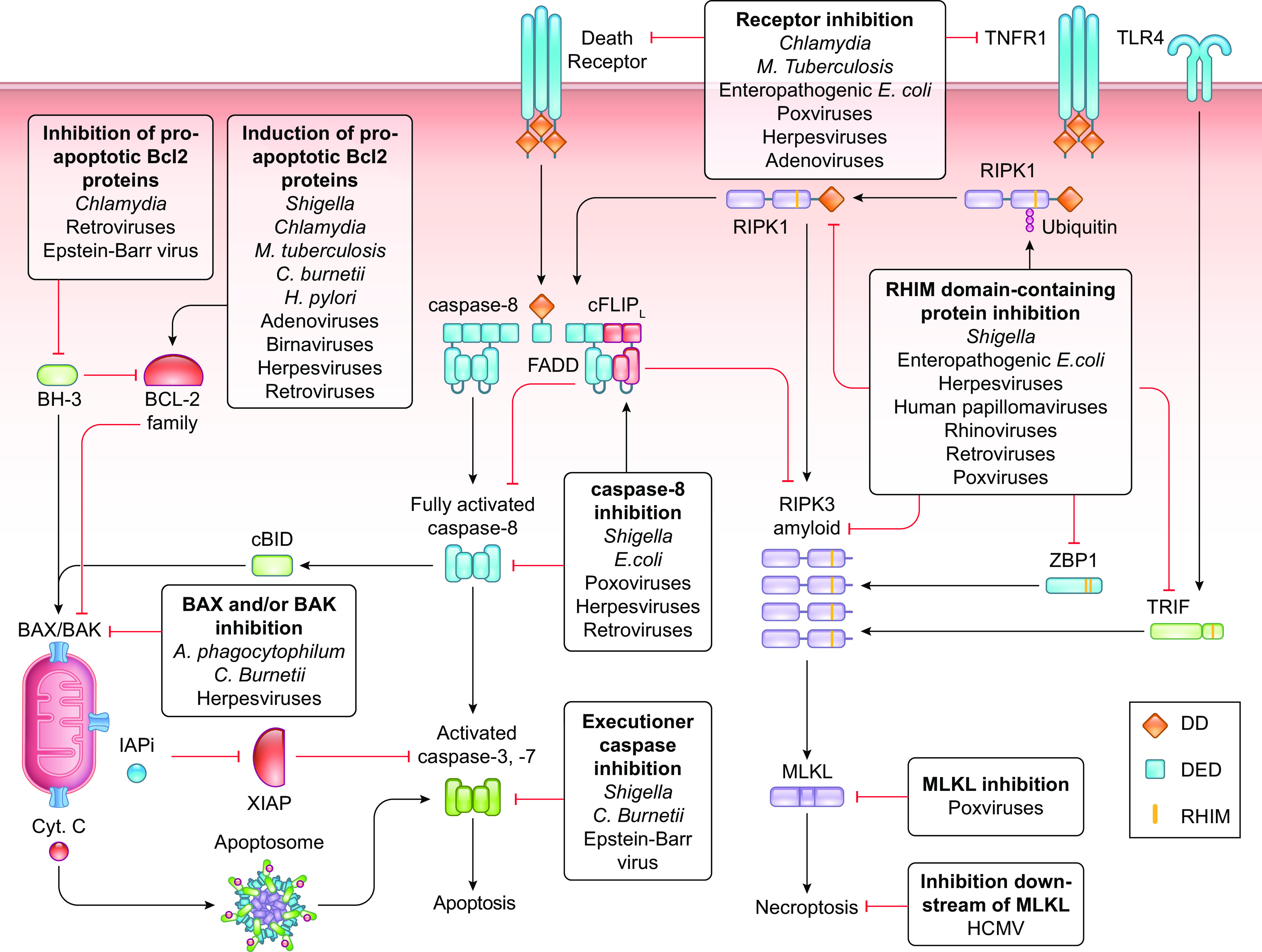
The coevolution of host-pathogen interactions underlies many human physiological traits associated with protection from or susceptibility to infections. Among the mechanisms that animals utilize to control infections are the regulated cell death pathways of pyroptosis, apoptosis, and necroptosis. Over the course of evolution these pathways have become intricate and complex, coevolving with microbes that infect animal hosts. Microbes, in turn, have evolved strategies to interfere with the pathways of regulated cell death to avoid eradication by the host. Here, we present an overview of the mechanisms of regulated cell death in Animalia and the strategies devised by pathogens to interfere with these processes. We review the molecular pathways of regulated cell death, their roles in infection, and how they are perturbed by viruses and bacteria, providing insights into the coevolution of host-pathogen interactions and cell death pathways.
Keywords: apoptosis; cell death; infection; necroptosis; pyroptosis.
Conflict of interest statement
D.R.G. consults for Ventus Therapeutics and Inzen Therapeutics. B.T. has no conflicts of interest, financial or otherwise, to disclose.
Figures

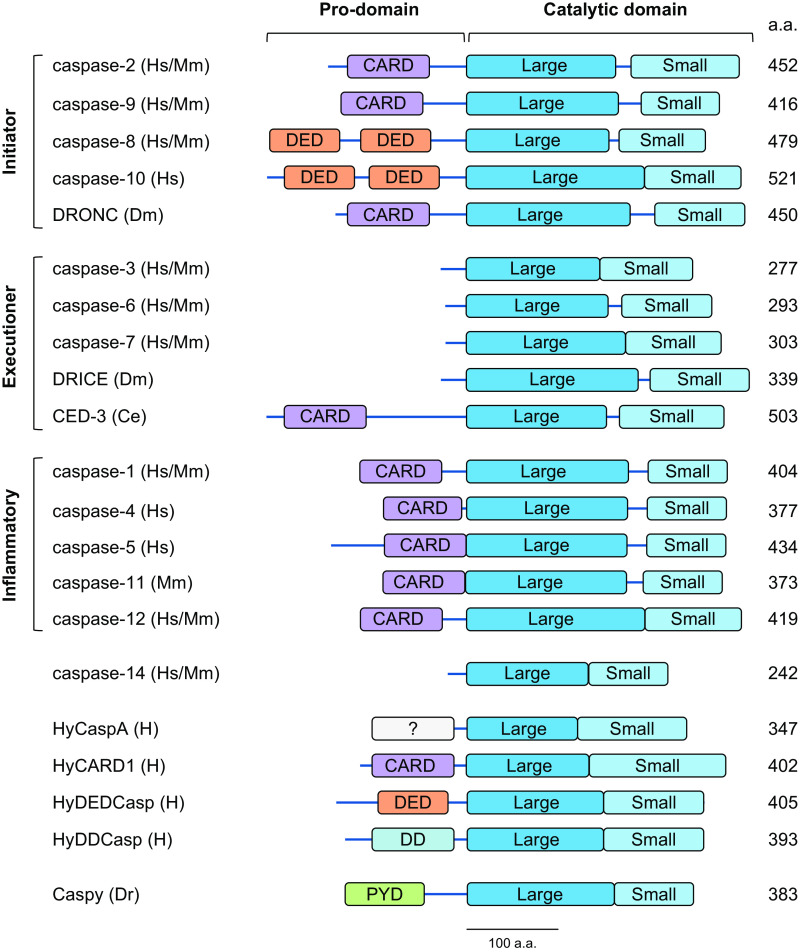
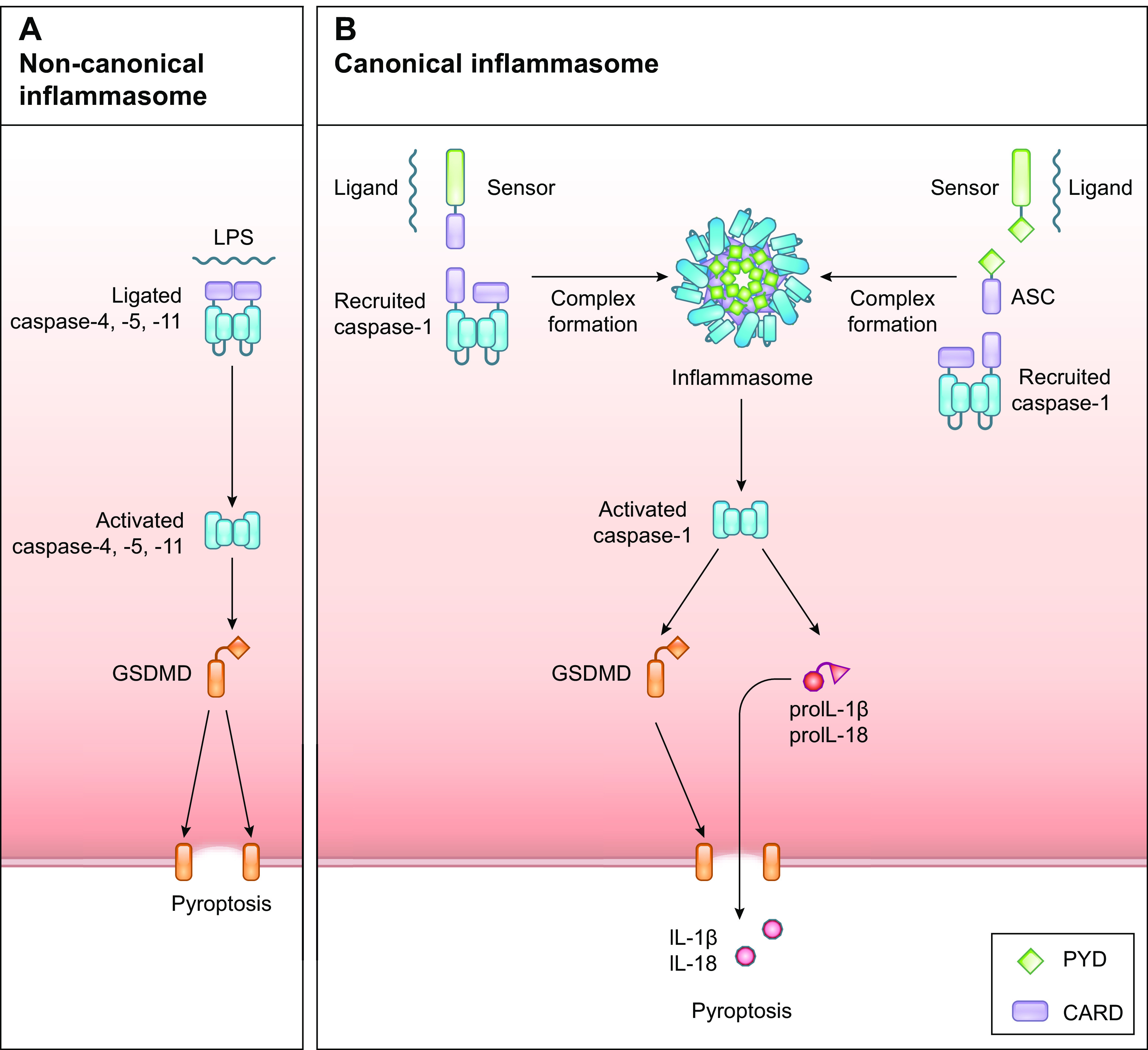
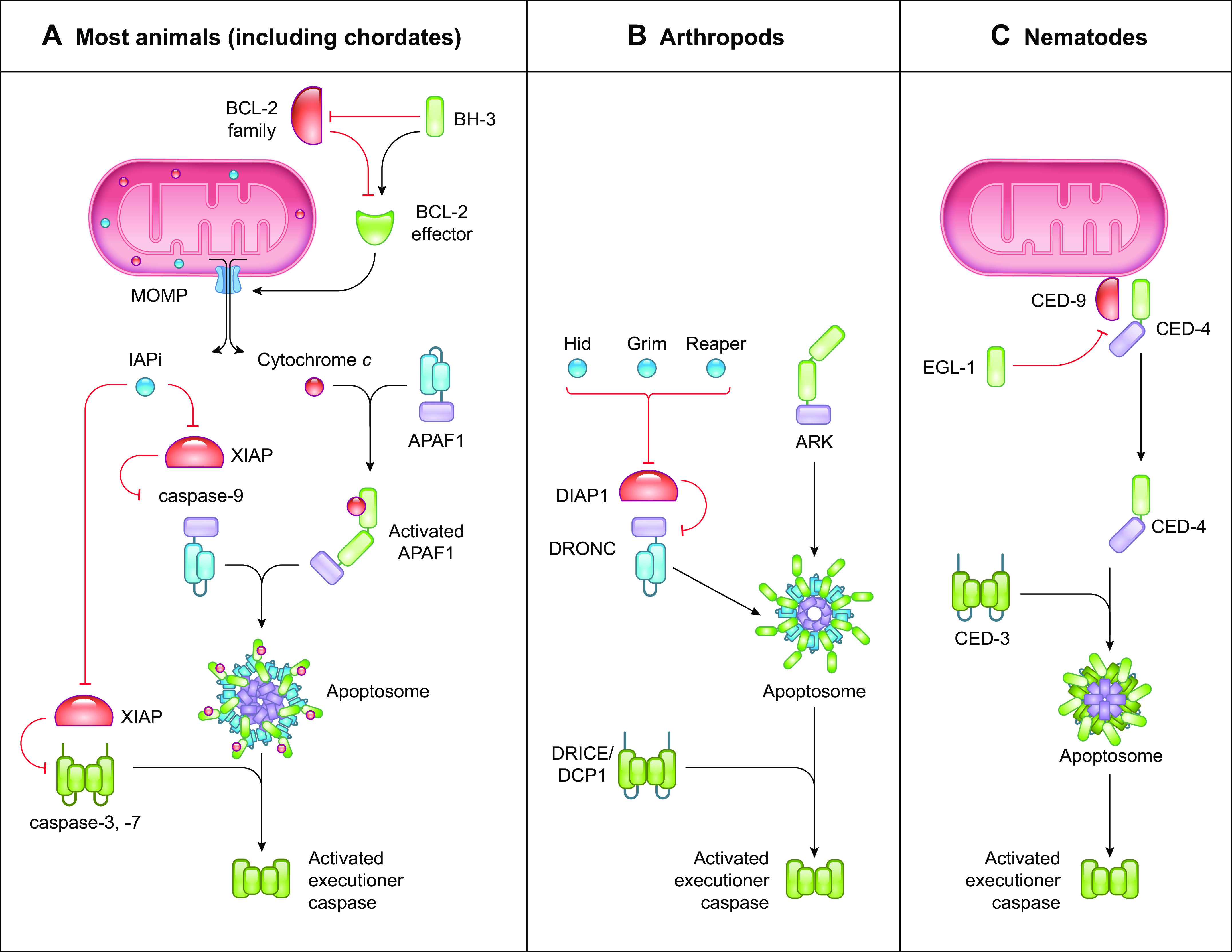



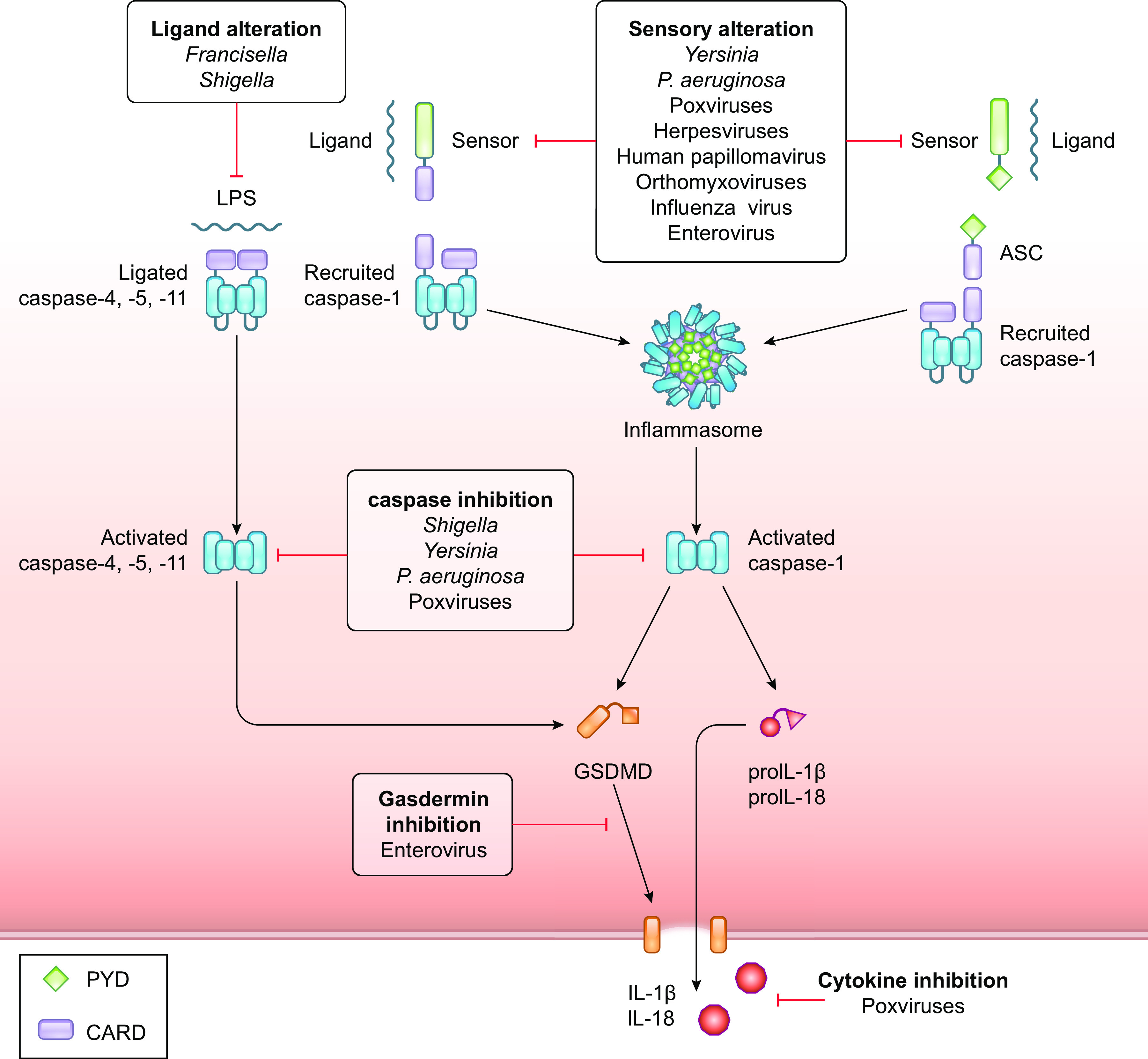
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources