

CoRE-ATAC: A deep learning model for the functional classification of regulatory elements from single cell and bulk ATAC-seq data
- PMID: 34898596
- PMCID: PMC8699717
- DOI: 10.1371/journal.pcbi.1009670
CoRE-ATAC: A deep learning model for the functional classification of regulatory elements from single cell and bulk ATAC-seq data
Abstract
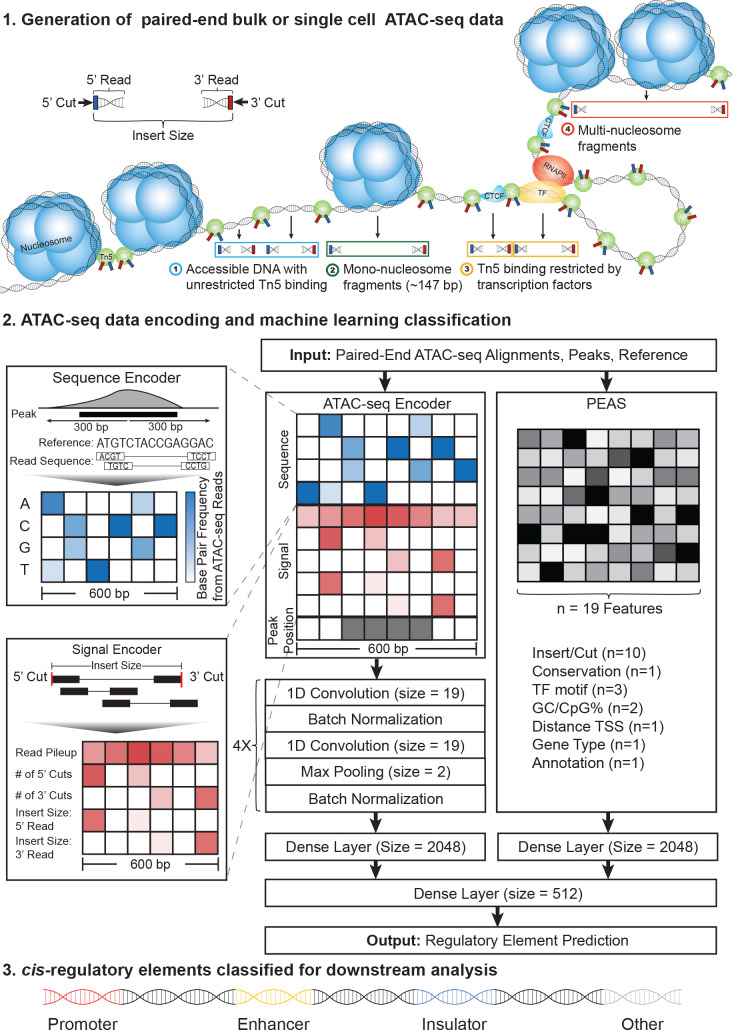
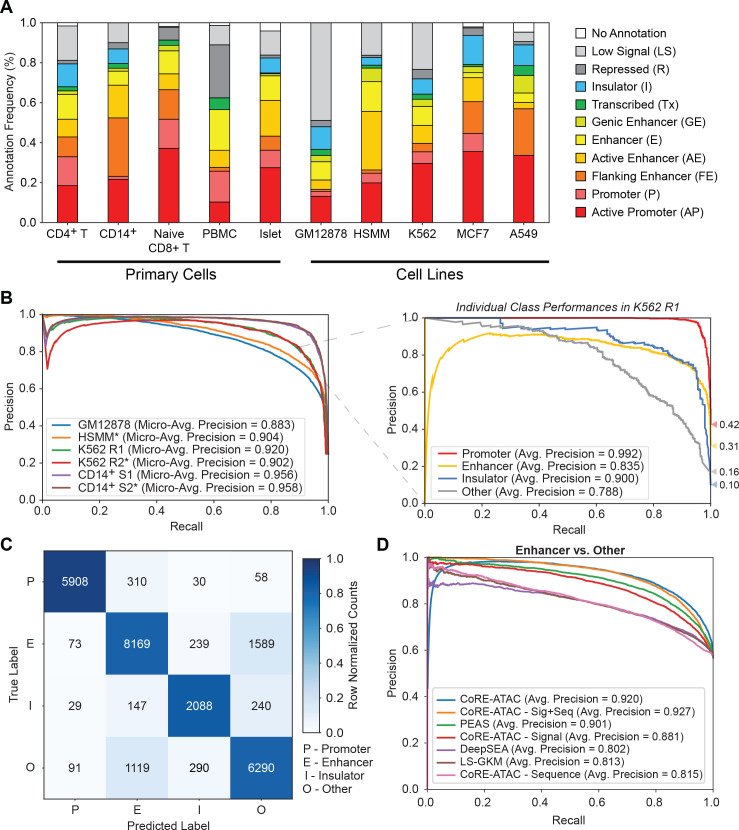
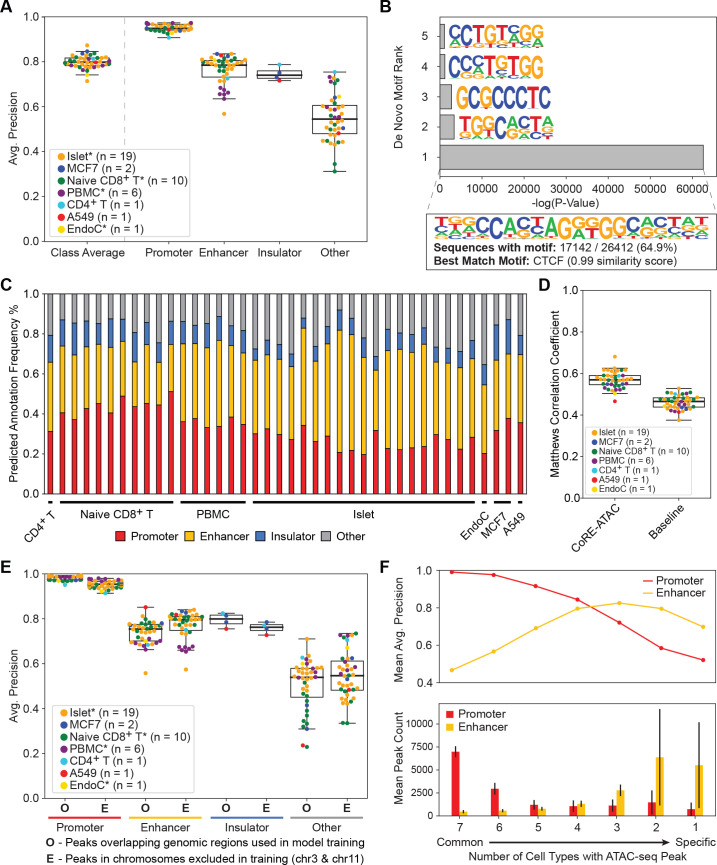
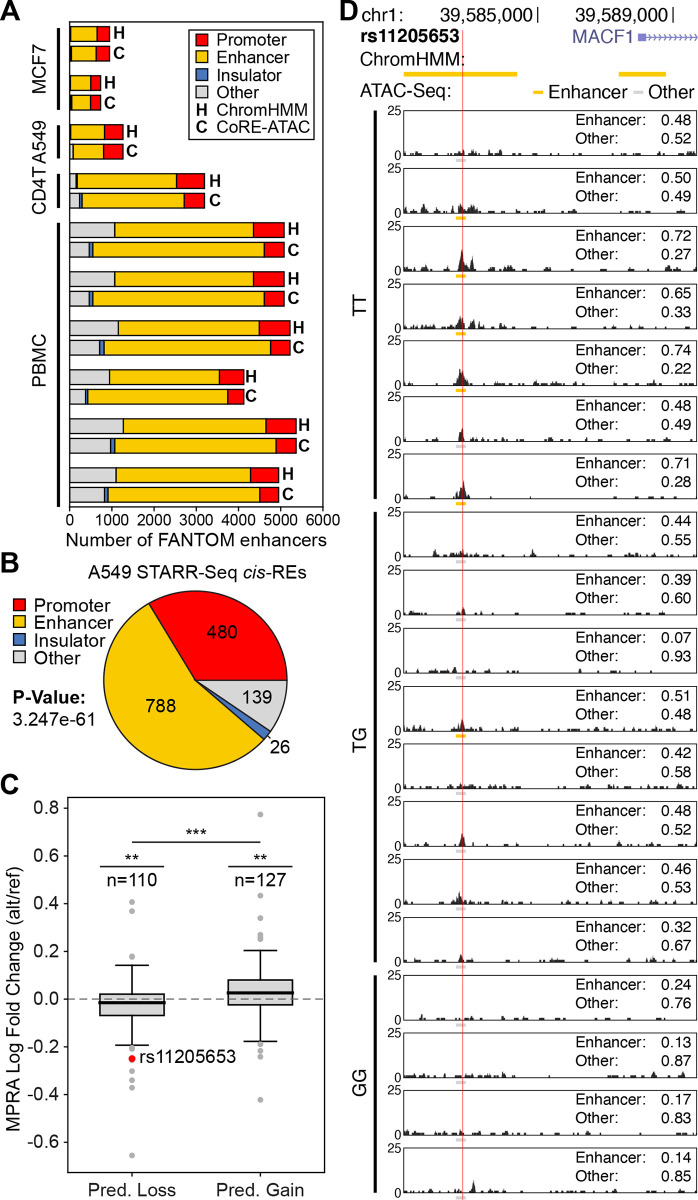
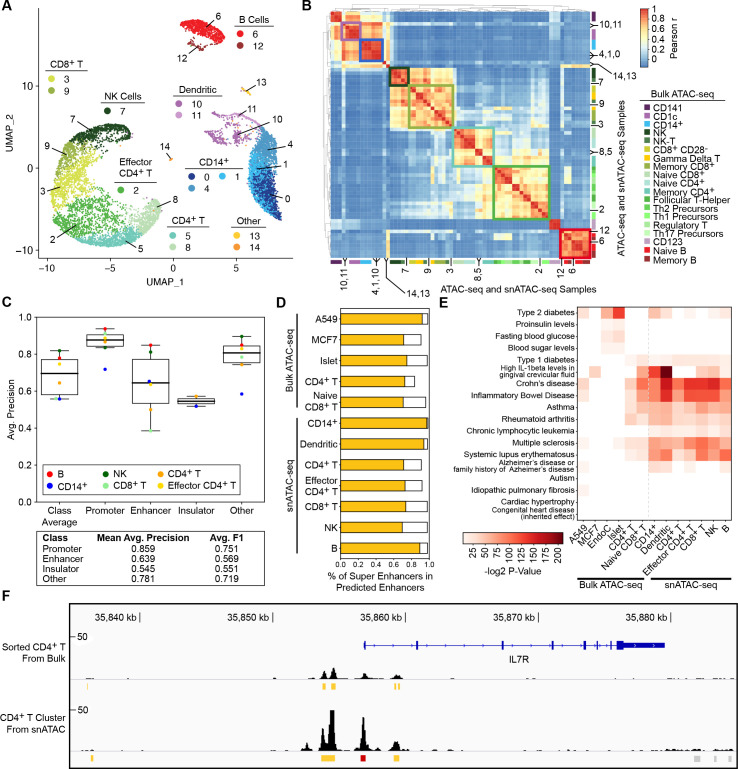
Cis-Regulatory elements (cis-REs) include promoters, enhancers, and insulators that regulate gene expression programs via binding of transcription factors. ATAC-seq technology effectively identifies active cis-REs in a given cell type (including from single cells) by mapping accessible chromatin at base-pair resolution. However, these maps are not immediately useful for inferring specific functions of cis-REs. For this purpose, we developed a deep learning framework (CoRE-ATAC) with novel data encoders that integrate DNA sequence (reference or personal genotypes) with ATAC-seq cut sites and read pileups. CoRE-ATAC was trained on 4 cell types (n = 6 samples/replicates) and accurately predicted known cis-RE functions from 7 cell types (n = 40 samples) that were not used in model training (mean average precision = 0.80, mean F1 score = 0.70). CoRE-ATAC enhancer predictions from 19 human islet samples coincided with genetically modulated gain/loss of enhancer activity, which was confirmed by massively parallel reporter assays (MPRAs). Finally, CoRE-ATAC effectively inferred cis-RE function from aggregate single nucleus ATAC-seq (snATAC) data from human blood-derived immune cells that overlapped with known functional annotations in sorted immune cells, which established the efficacy of these models to study cis-RE functions of rare cells without the need for cell sorting. ATAC-seq maps from primary human cells reveal individual- and cell-specific variation in cis-RE activity. CoRE-ATAC increases the functional resolution of these maps, a critical step for studying regulatory disruptions behind diseases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences. 2009. Jun 9;106(23):9362–7. doi: 10.1073/pnas.0903103106 - DOI - PMC - PubMed
-
- Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proceedings of the National Academy of Sciences. 2013. Oct 29;110(44):17921–6. doi: 10.1073/pnas.1317023110 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
