

Fungal diversity notes 1387-1511: taxonomic and phylogenetic contributions on genera and species of fungal taxa
- PMID: 34899100
- PMCID: PMC8648402
- DOI: 10.1007/s13225-021-00489-3
Fungal diversity notes 1387-1511: taxonomic and phylogenetic contributions on genera and species of fungal taxa
Abstract
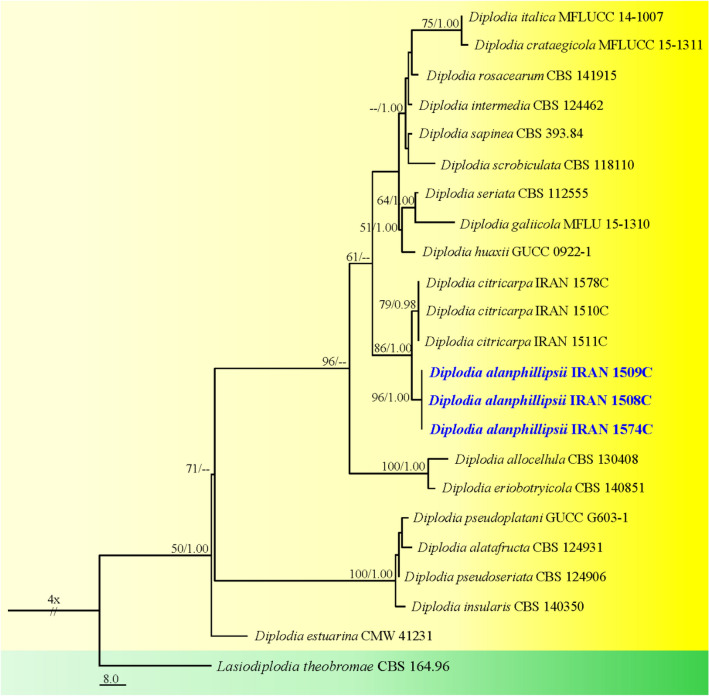
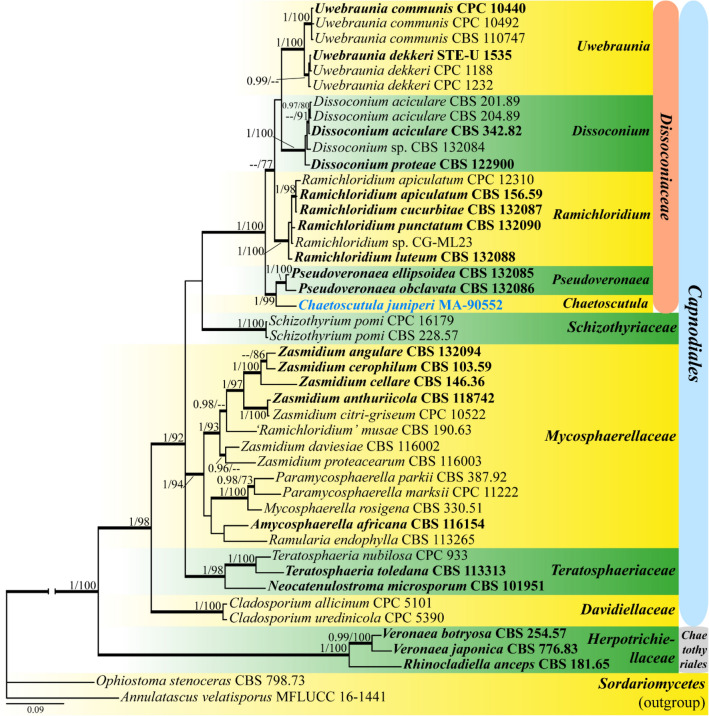
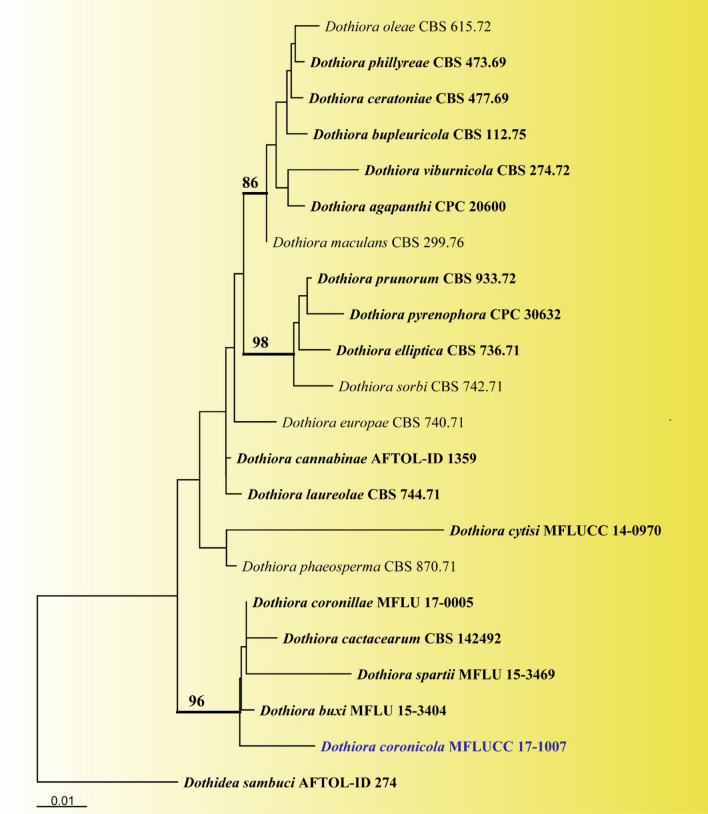
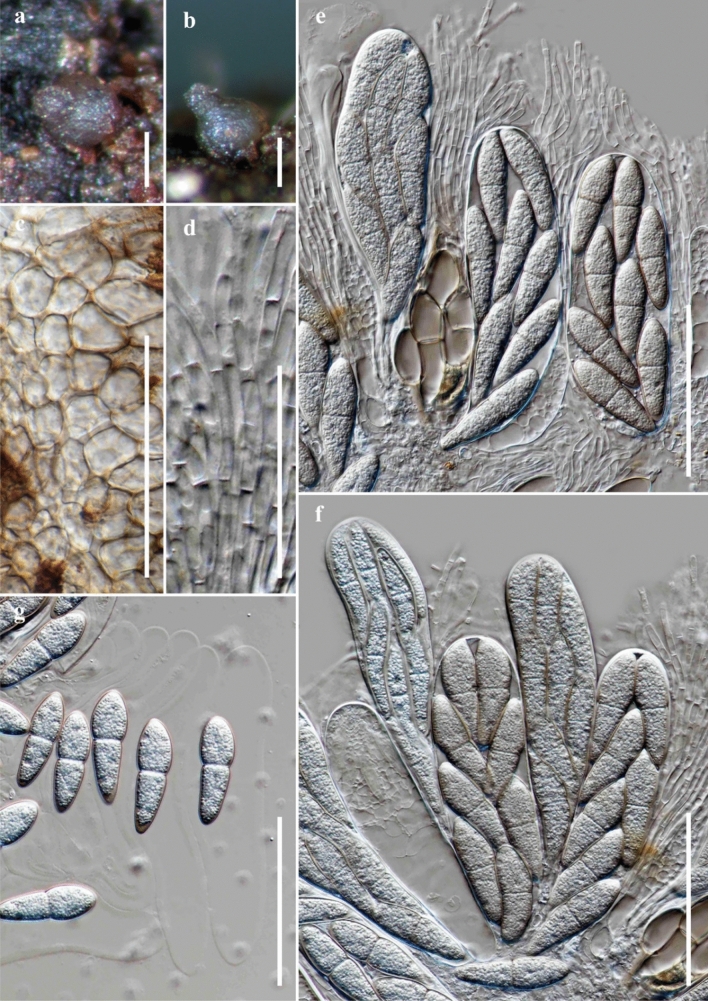
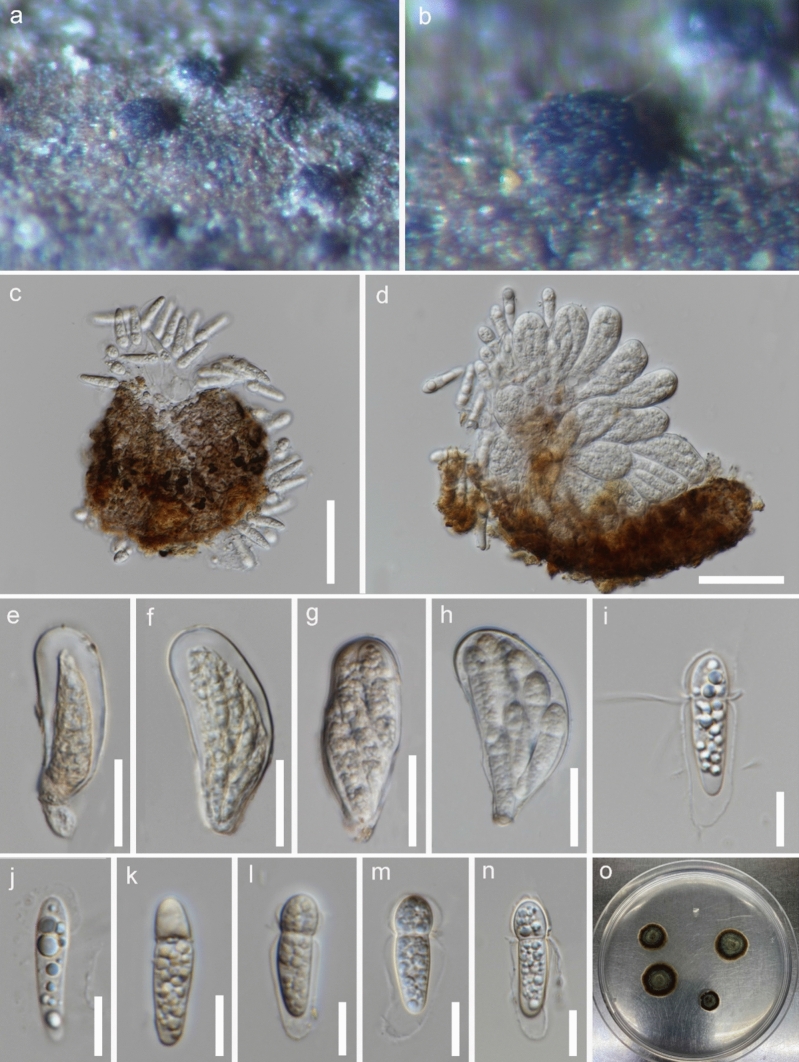
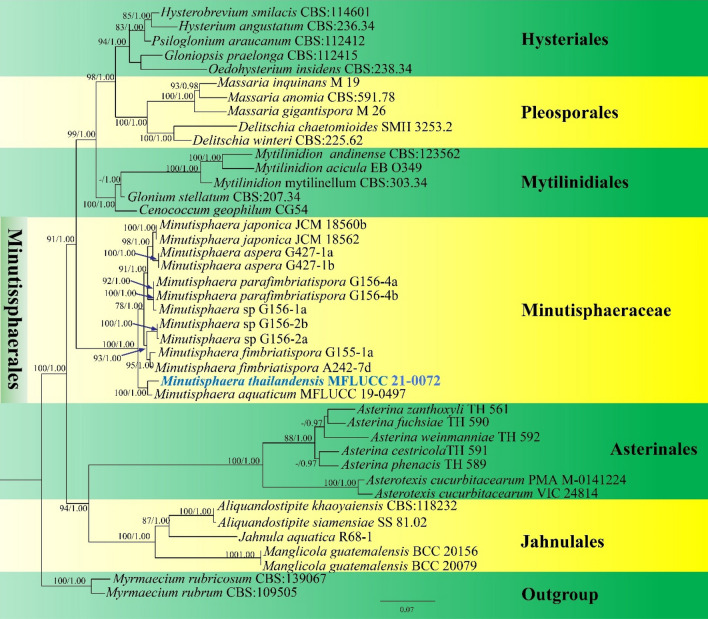
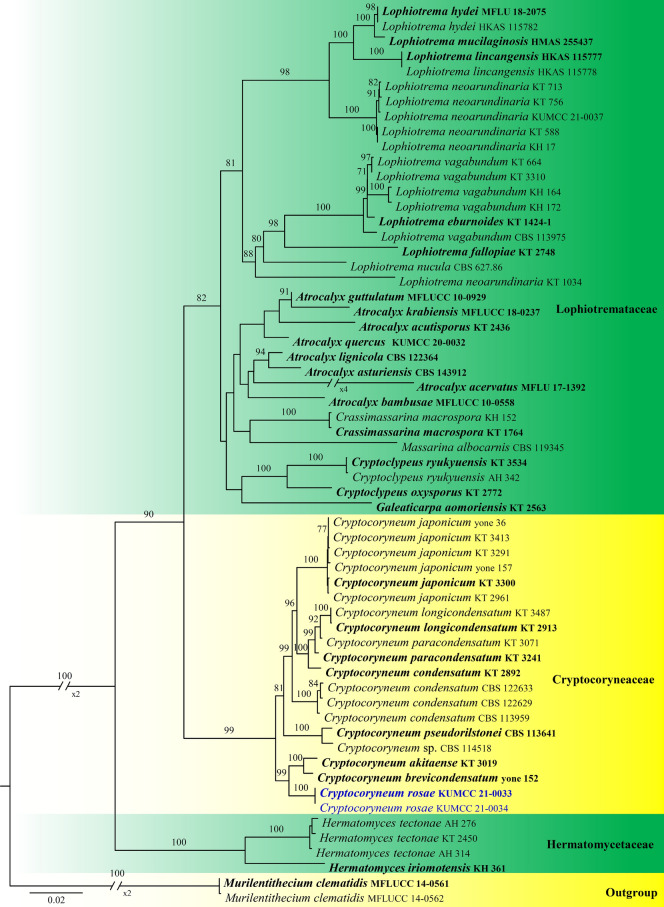
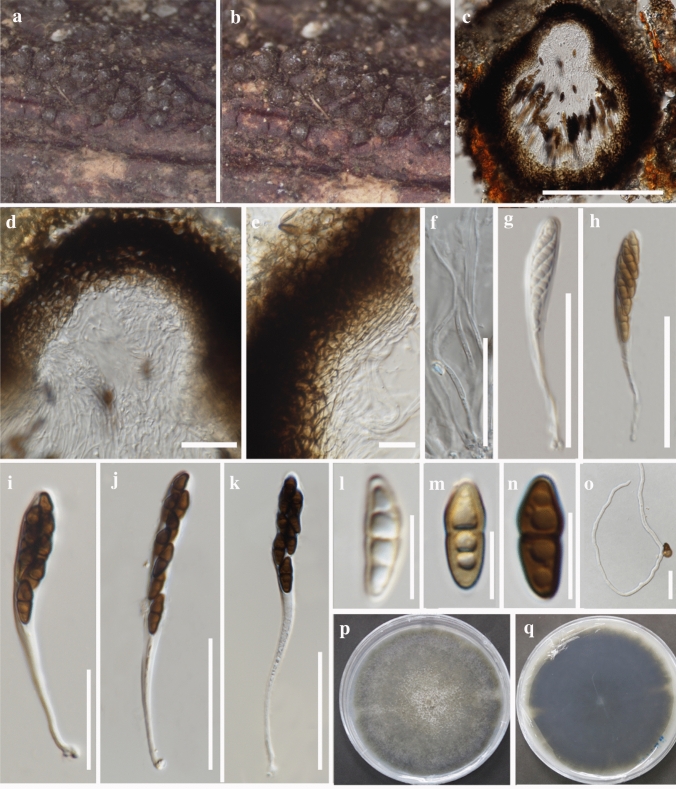
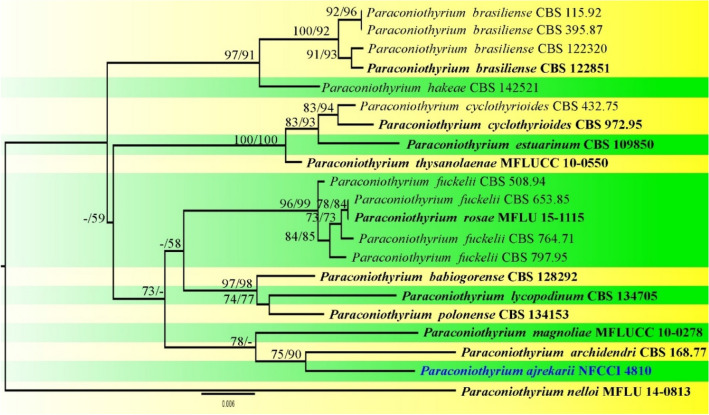
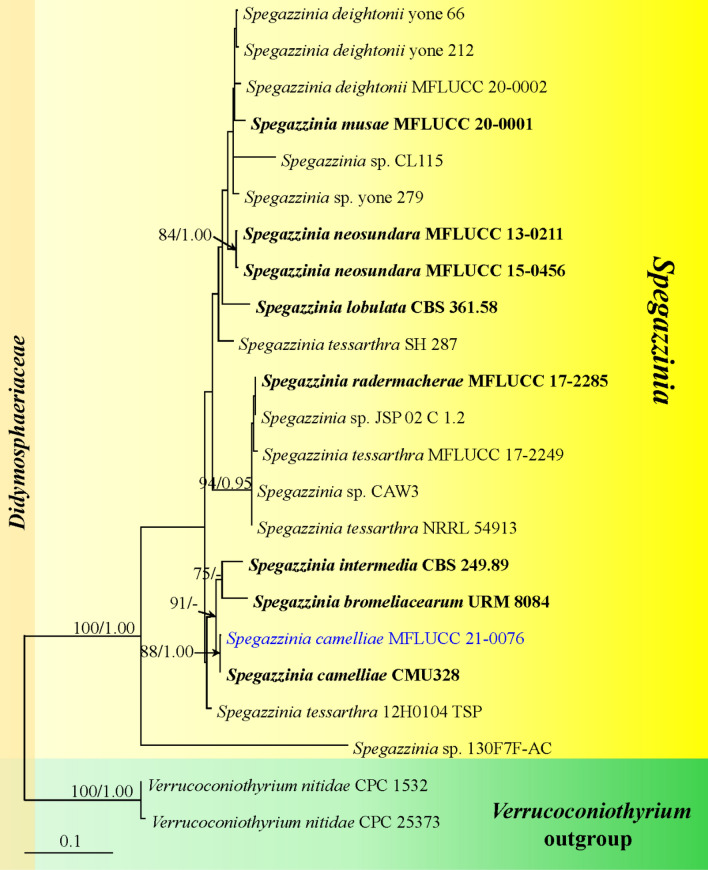
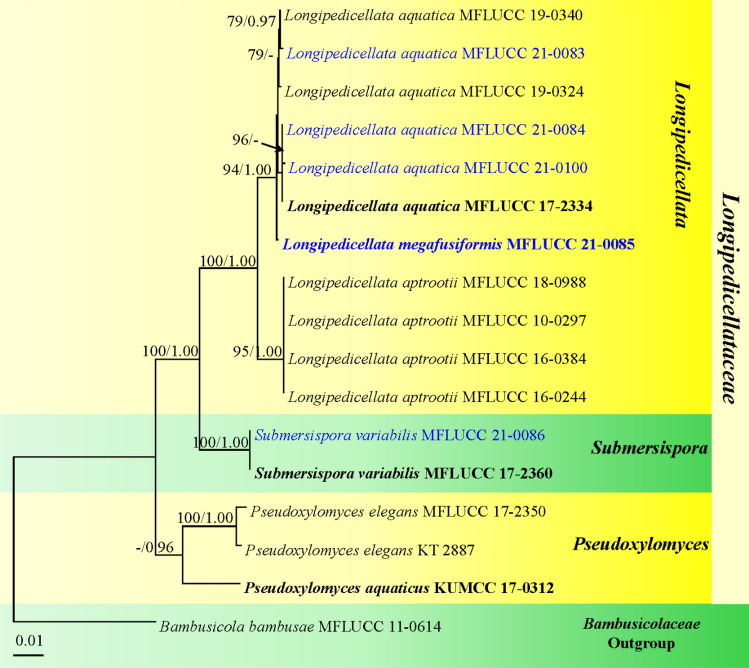
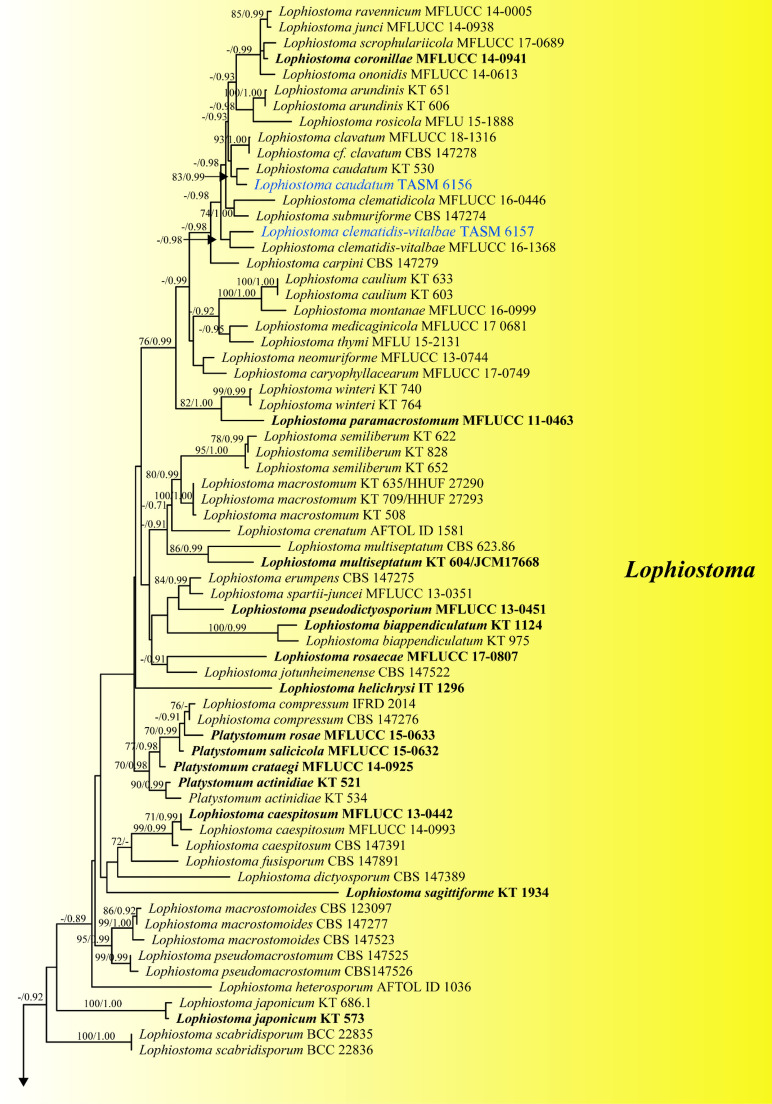
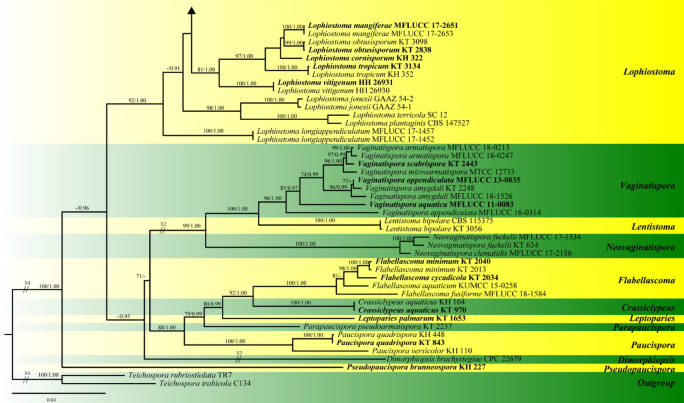
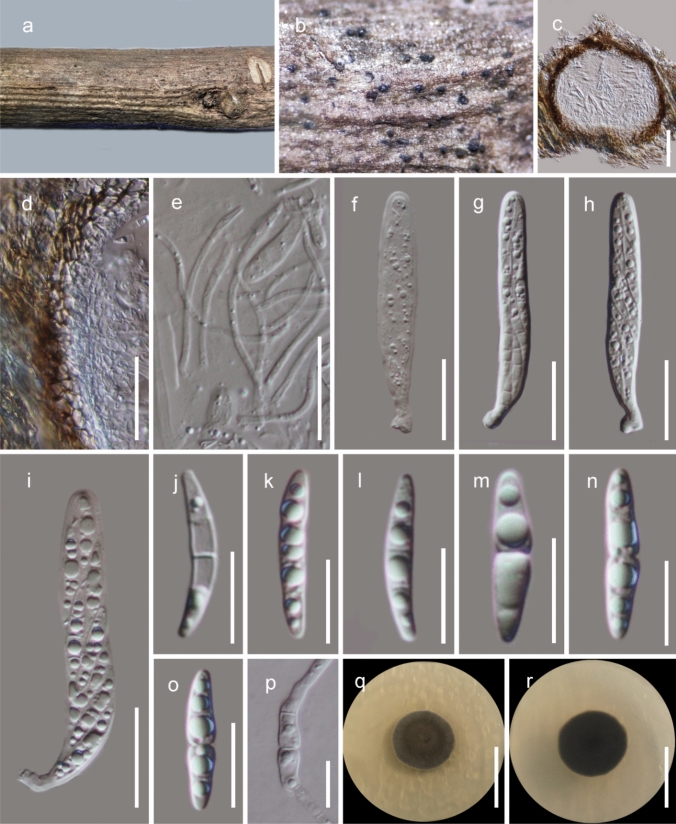
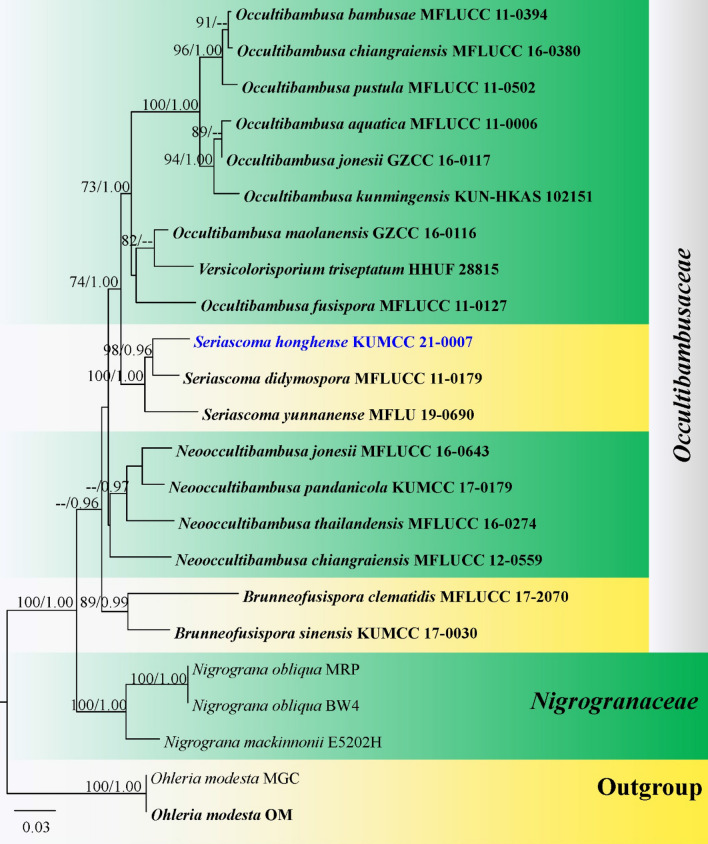
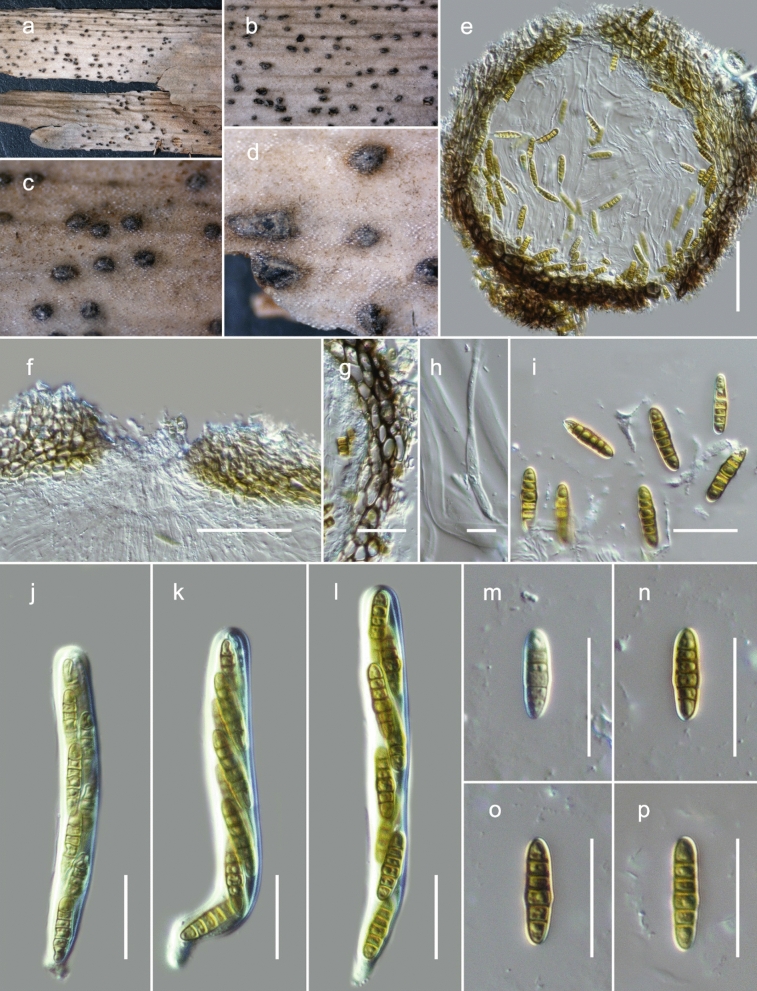
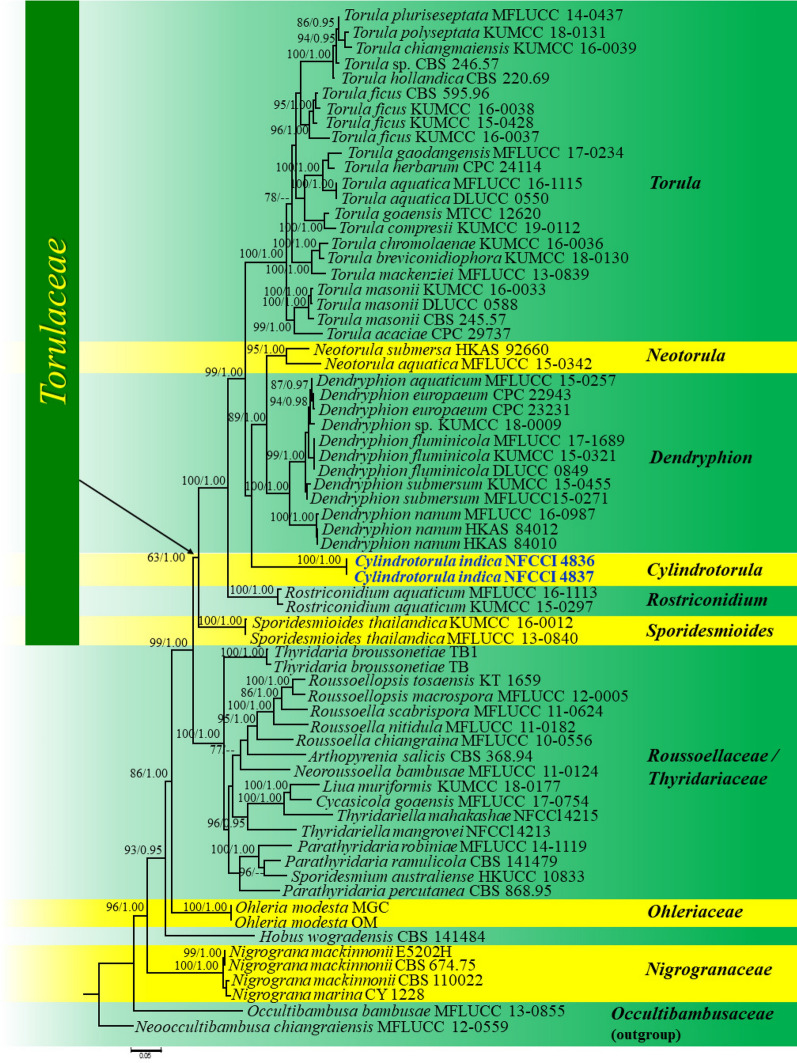
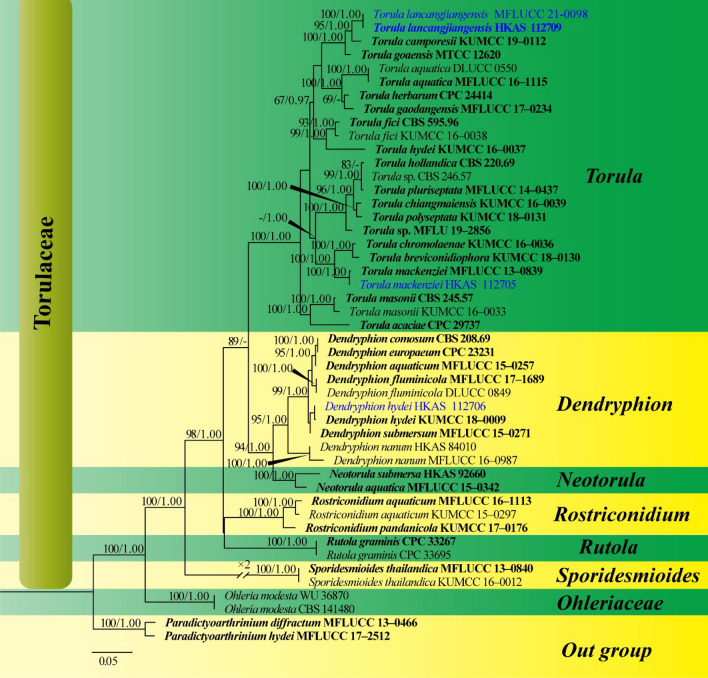
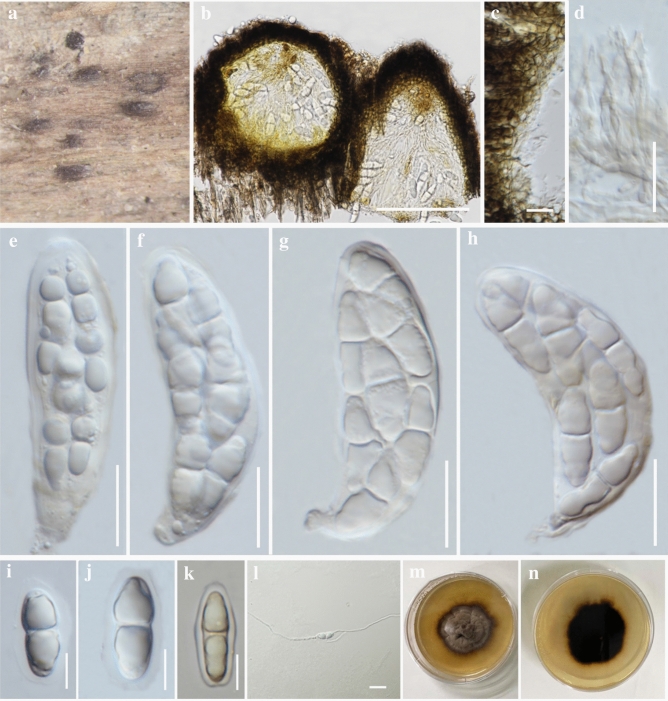
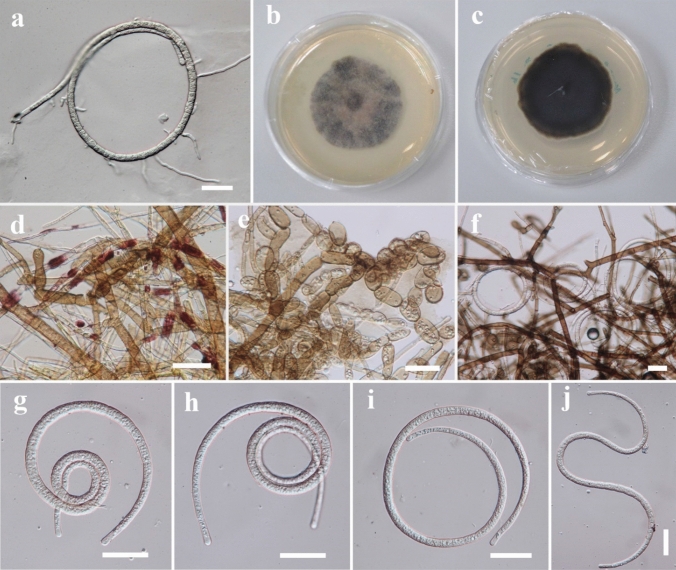
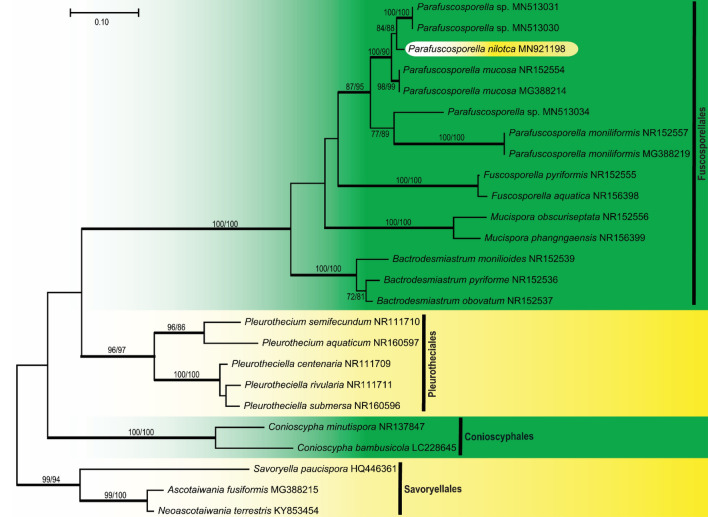
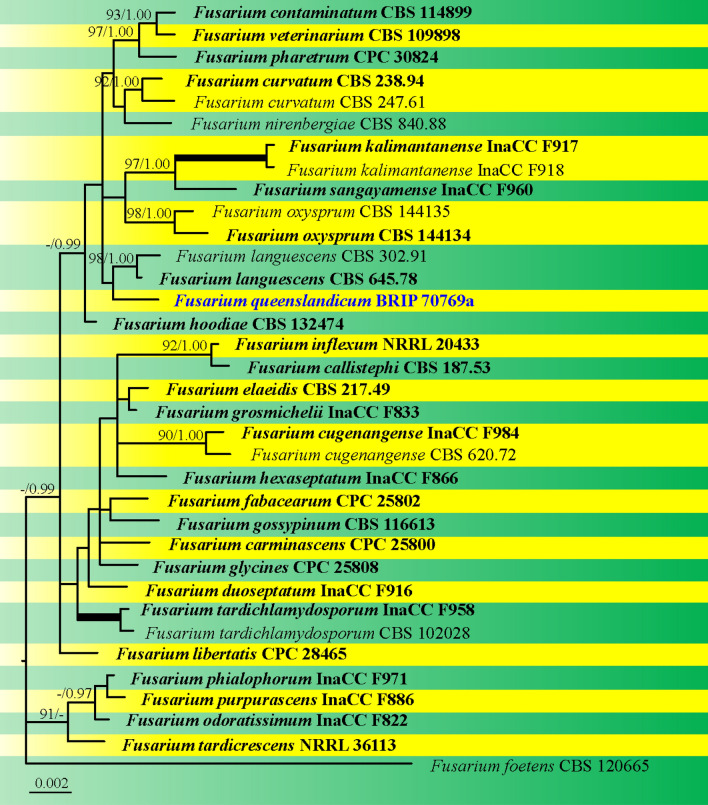
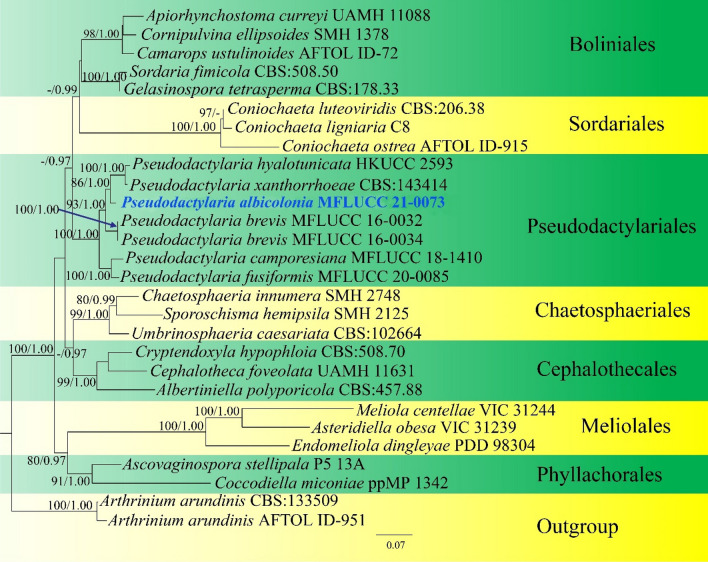
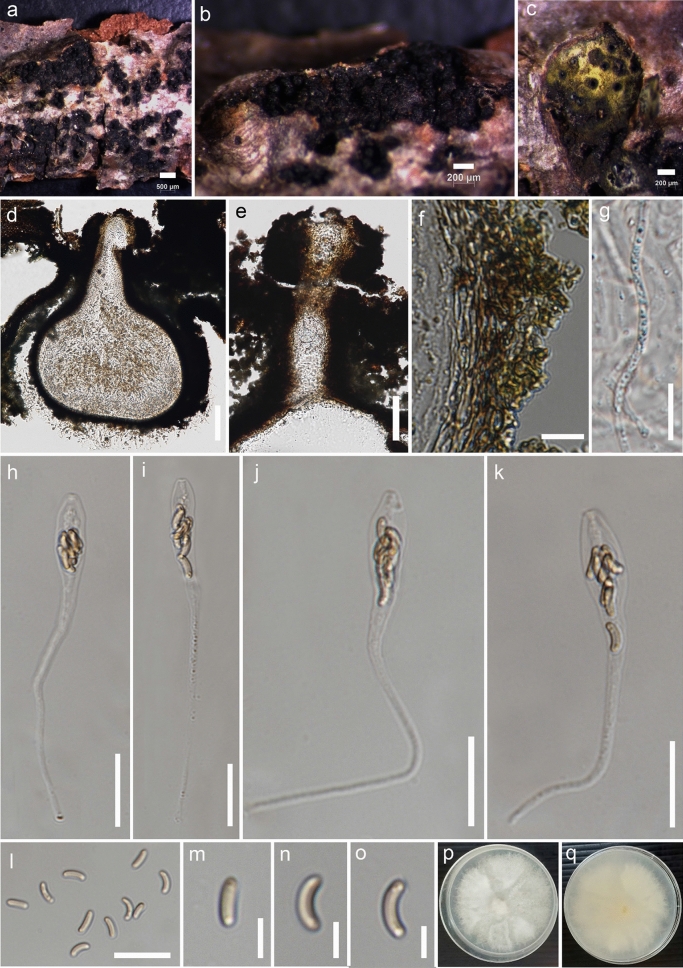
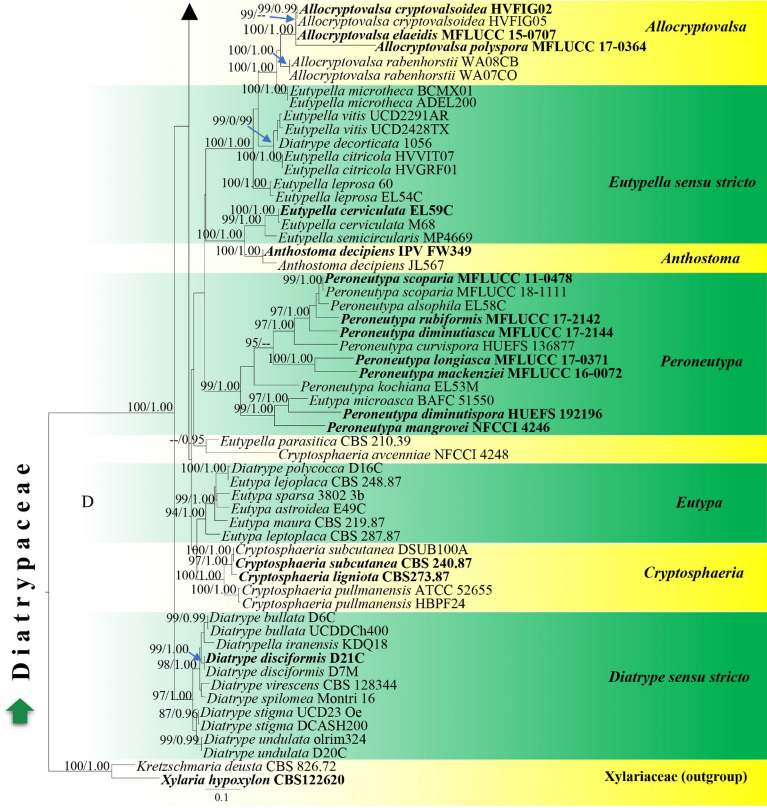
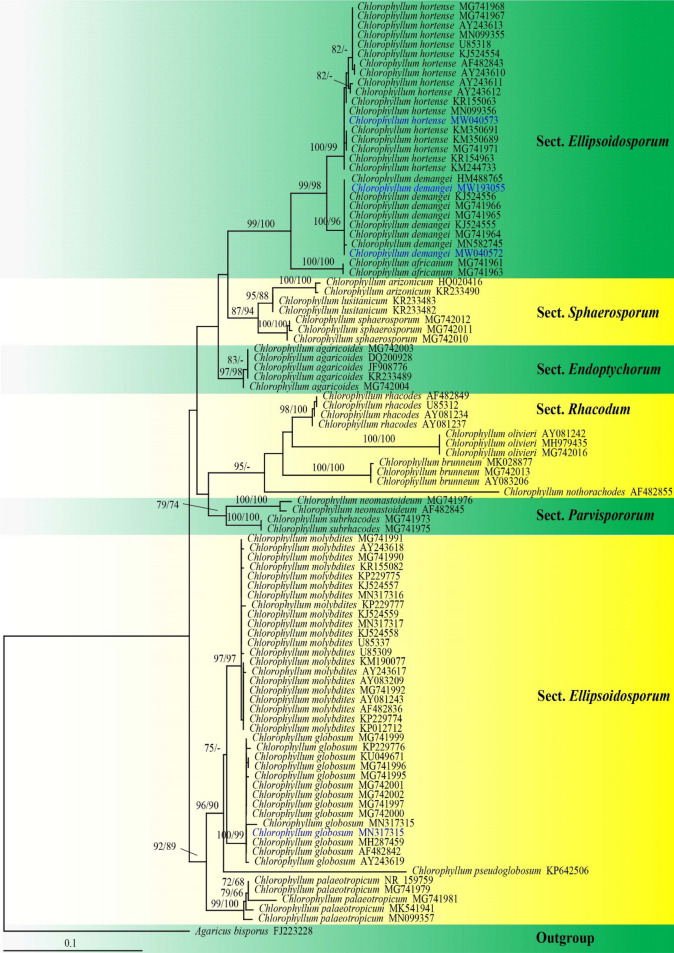
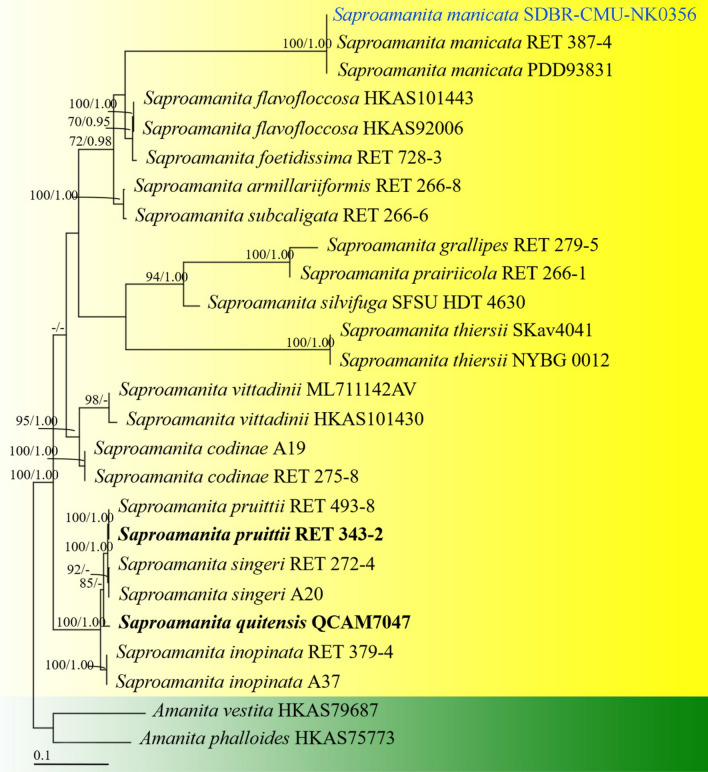
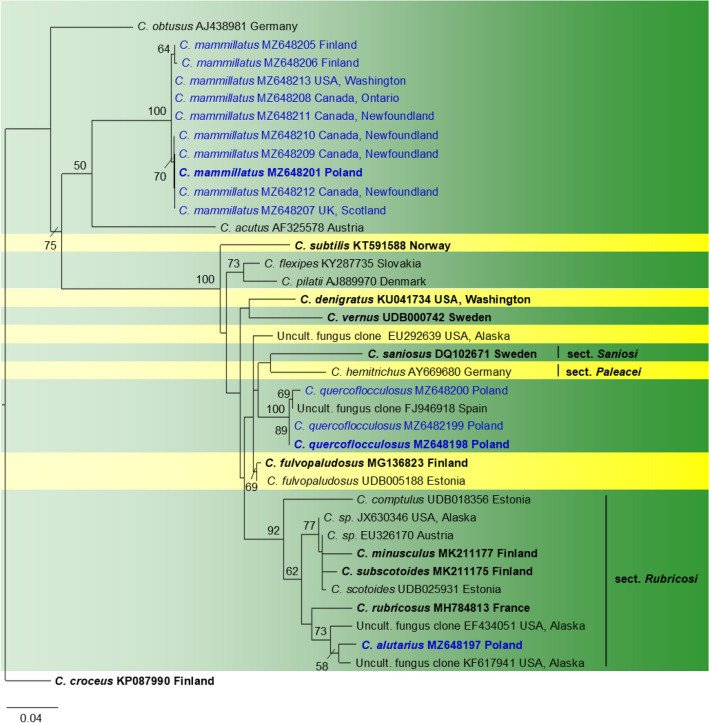
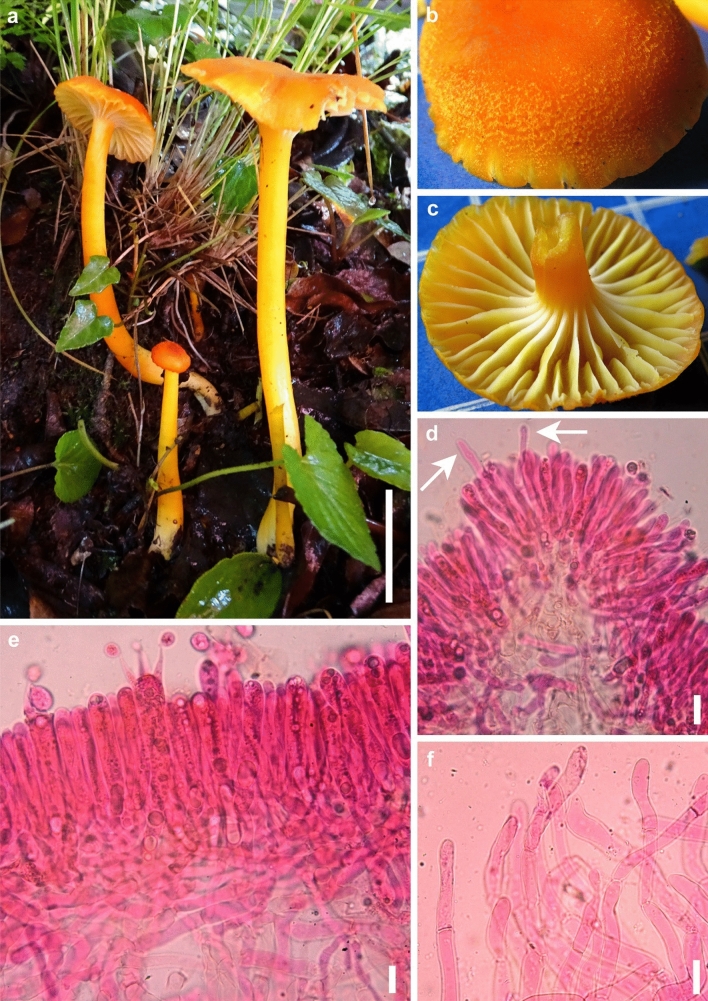
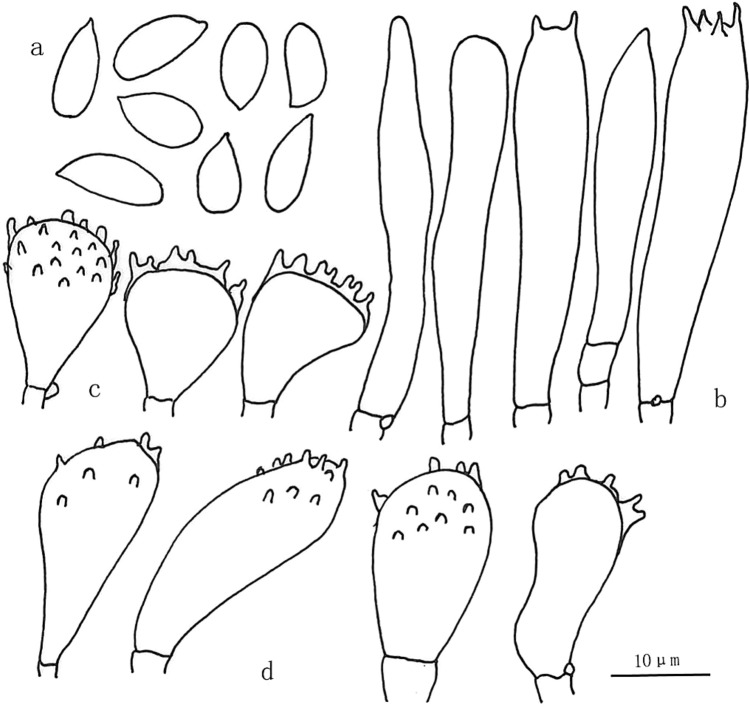
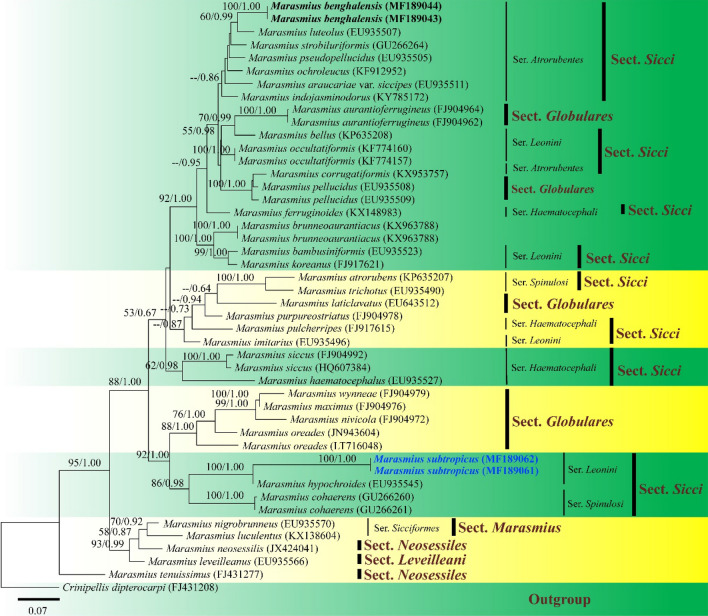
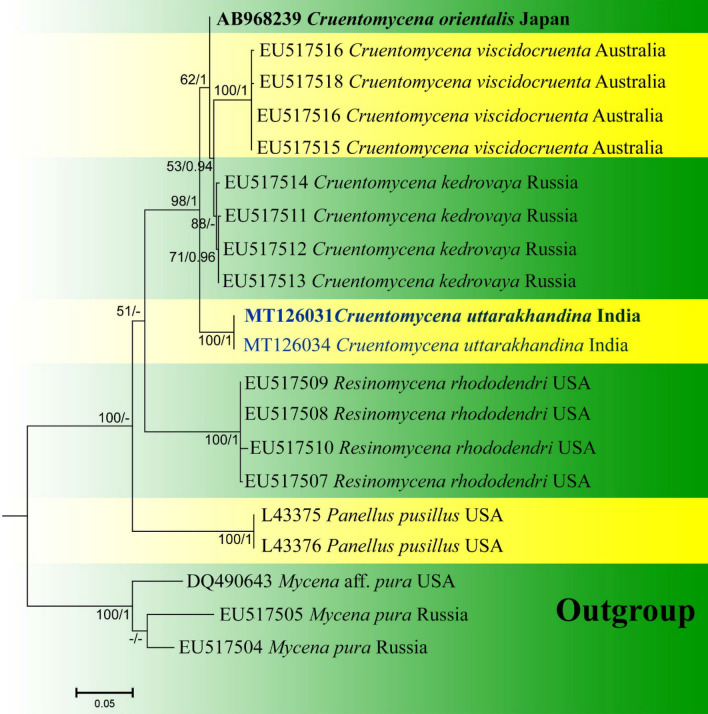
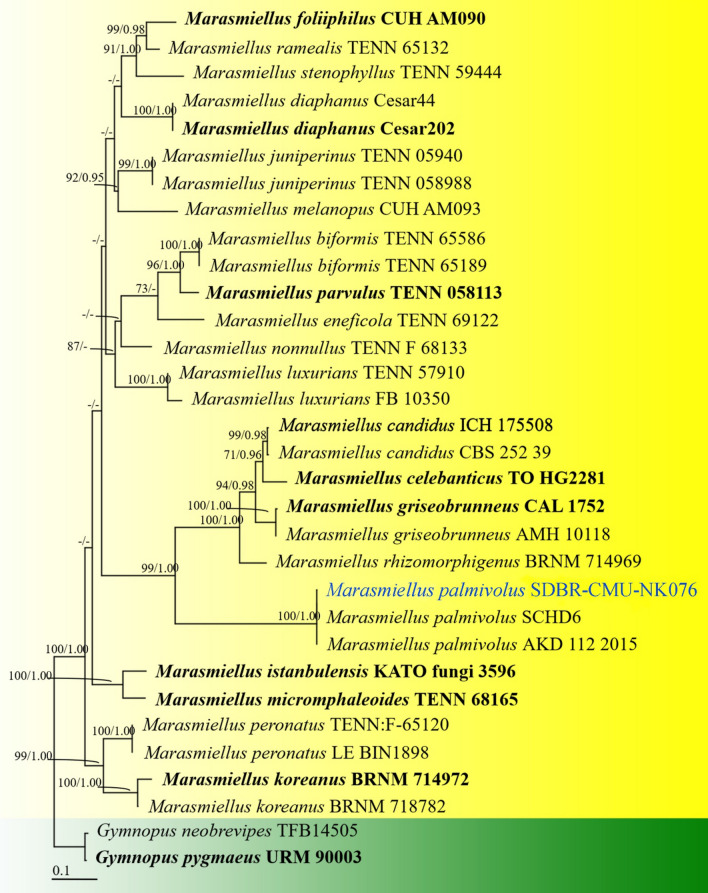
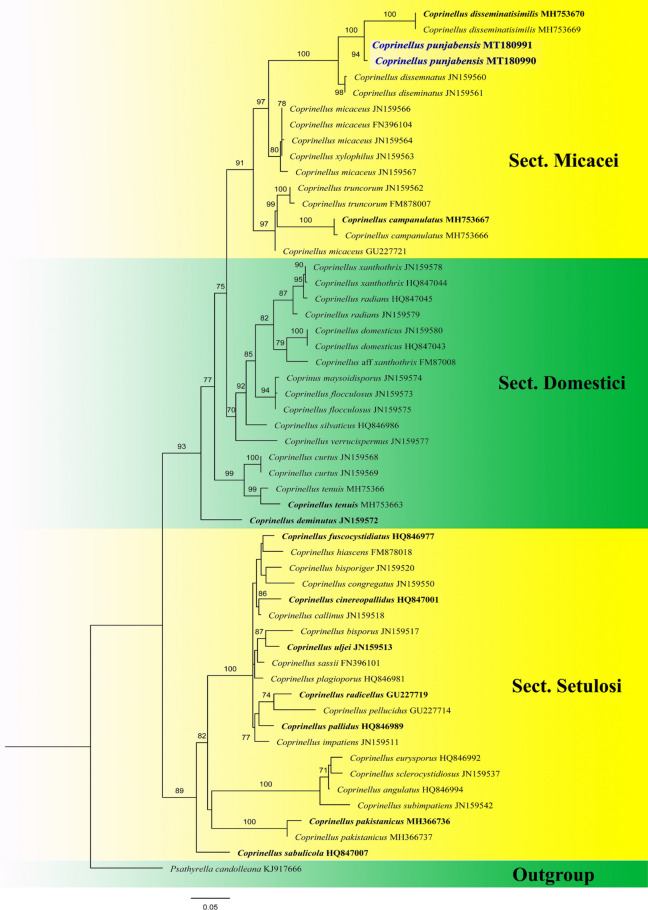
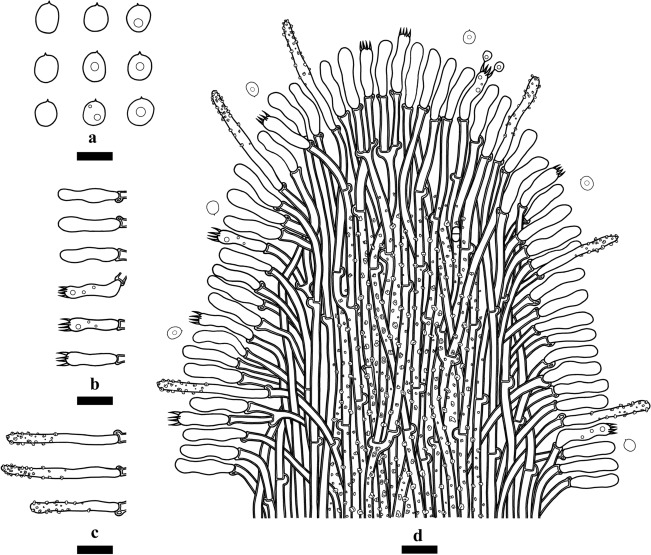
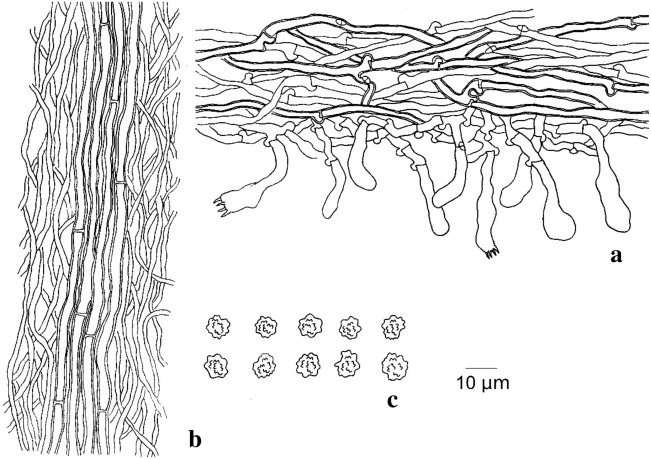
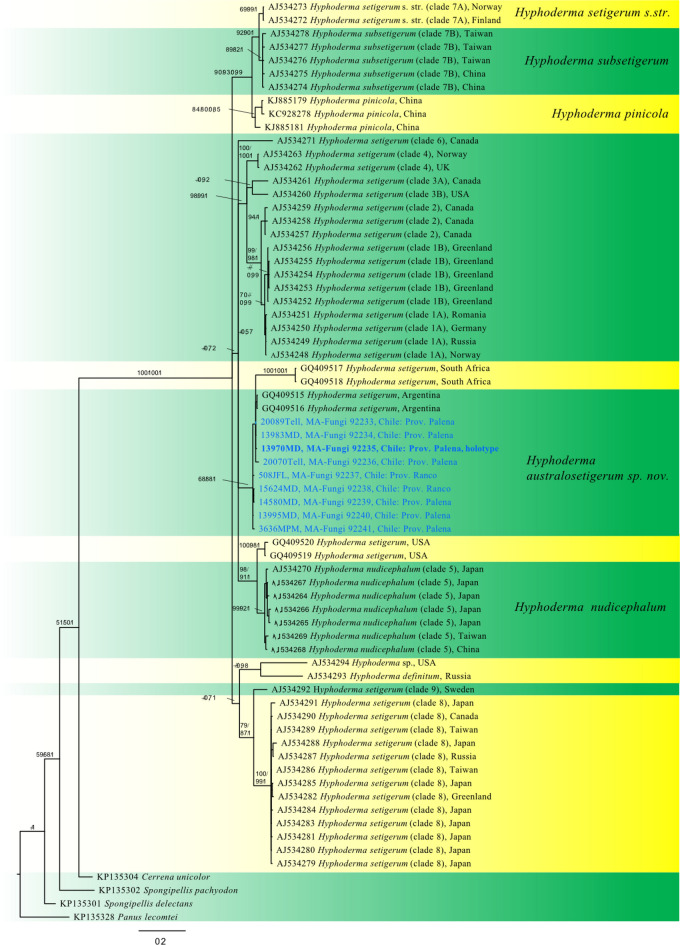
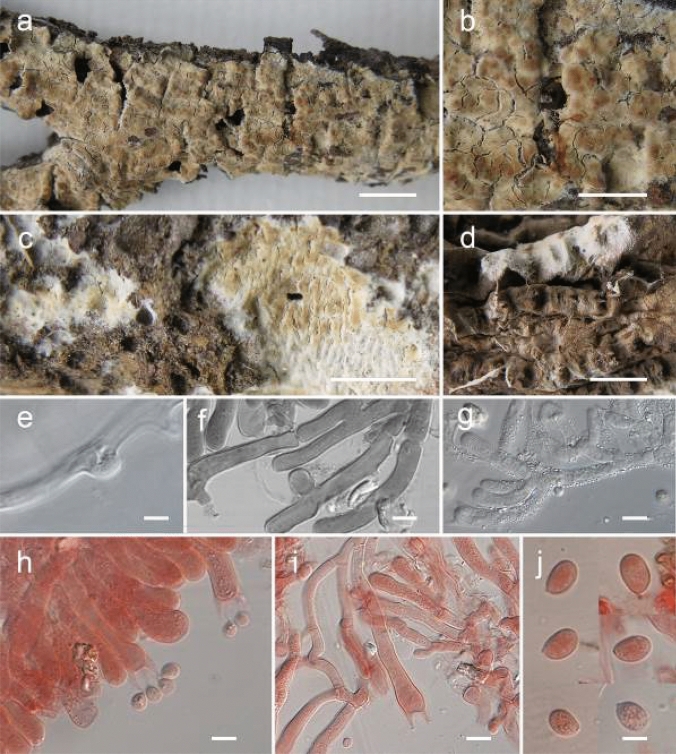
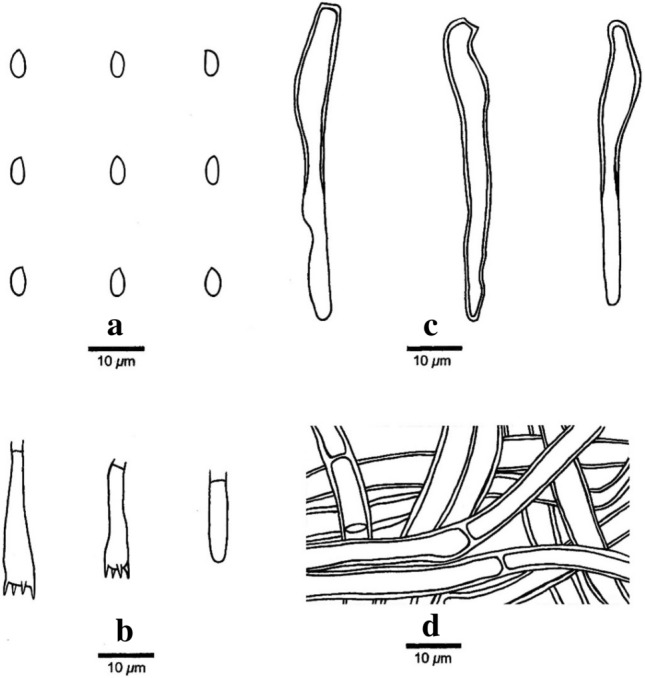
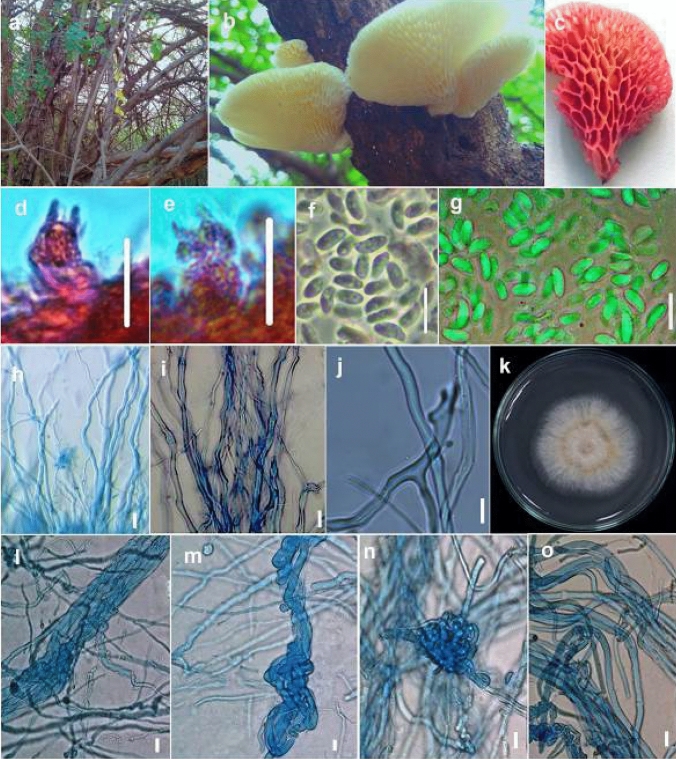
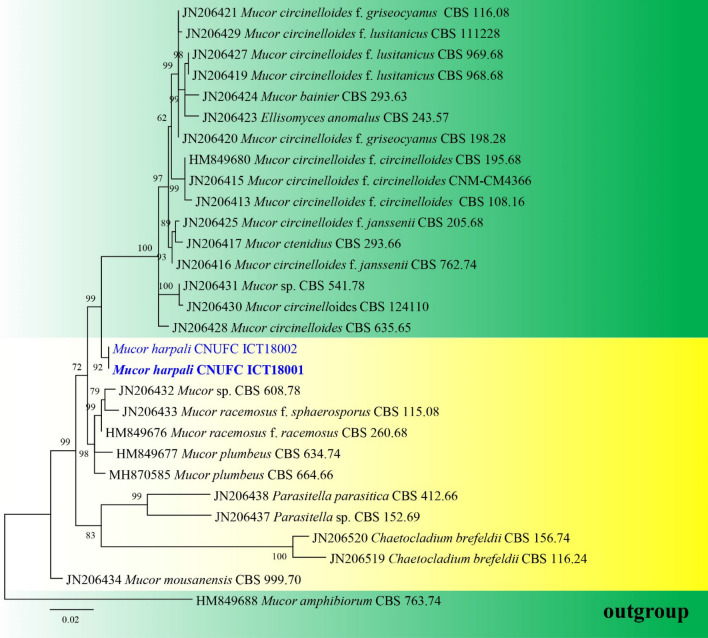
This article is the 13th contribution in the Fungal Diversity Notes series, wherein 125 taxa from four phyla, ten classes, 31 orders, 69 families, 92 genera and three genera incertae sedis are treated, demonstrating worldwide and geographic distribution. Fungal taxa described and illustrated in the present study include three new genera, 69 new species, one new combination, one reference specimen and 51 new records on new hosts and new geographical distributions. Three new genera, Cylindrotorula (Torulaceae), Scolecoleotia (Leotiales genus incertae sedis) and Xenovaginatispora (Lindomycetaceae) are introduced based on distinct phylogenetic lineages and unique morphologies. Newly described species are Aspergillus lannaensis, Cercophora dulciaquae, Cladophialophora aquatica, Coprinellus punjabensis, Cortinarius alutarius, C. mammillatus, C. quercoflocculosus, Coryneum fagi, Cruentomycena uttarakhandina, Cryptocoryneum rosae, Cyathus uniperidiolus, Cylindrotorula indica, Diaporthe chamaeropicola, Didymella azollae, Diplodia alanphillipsii, Dothiora coronicola, Efibula rodriguezarmasiae, Erysiphe salicicola, Fusarium queenslandicum, Geastrum gorgonicum, G. hansagiense, Helicosporium sexualis, Helminthosporium chiangraiensis, Hongkongmyces kokensis, Hydrophilomyces hydraenae, Hygrocybe boertmannii, Hyphoderma australosetigerum, Hyphodontia yunnanensis, Khaleijomyces umikazeana, Laboulbenia divisa, Laboulbenia triarthronis, Laccaria populina, Lactarius pallidozonarius, Lepidosphaeria strobelii, Longipedicellata megafusiformis, Lophiotrema lincangensis, Marasmius benghalensis, M. jinfoshanensis, M. subtropicus, Mariannaea camelliae, Melanographium smilaxii, Microbotryum polycnemoides, Mimeomyces digitatus, Minutisphaera thailandensis, Mortierella solitaria, Mucor harpali, Nigrograna jinghongensis, Odontia huanrenensis, O. parvispina, Paraconiothyrium ajrekarii, Parafuscosporella niloticus, Phaeocytostroma yomensis, Phaeoisaria synnematicus, Phanerochaete hainanensis, Pleopunctum thailandicum, Pleurotheciella dimorphospora, Pseudochaetosphaeronema chiangraiense, Pseudodactylaria albicolonia, Rhexoacrodictys nigrospora, Russula paravioleipes, Scolecoleotia eriocamporesi, Seriascoma honghense, Synandromyces makranczyi, Thyridaria aureobrunnea, Torula lancangjiangensis, Tubeufia longihelicospora, Wicklowia fusiformispora, Xenovaginatispora phichaiensis and Xylaria apiospora. One new combination, Pseudobactrodesmium stilboideus is proposed. A reference specimen of Comoclathris permunda is designated. New host or distribution records are provided for Acrocalymma fici, Aliquandostipite khaoyaiensis, Camarosporidiella laburni, Canalisporium caribense, Chaetoscutula juniperi, Chlorophyllum demangei, C. globosum, C. hortense, Cladophialophora abundans, Dendryphion hydei, Diaporthe foeniculina, D. pseudophoenicicola, D. pyracanthae, Dictyosporium pandanicola, Dyfrolomyces distoseptatus, Ernakulamia tanakae, Eutypa flavovirens, E. lata, Favolus septatus, Fusarium atrovinosum, F. clavum, Helicosporium luteosporum, Hermatomyces nabanheensis, Hermatomyces sphaericoides, Longipedicellata aquatica, Lophiostoma caudata, L. clematidis-vitalbae, Lophiotrema hydei, L. neoarundinaria, Marasmiellus palmivorus, Megacapitula villosa, Micropsalliota globocystis, M. gracilis, Montagnula thailandica, Neohelicosporium irregulare, N. parisporum, Paradictyoarthrinium diffractum, Phaeoisaria aquatica, Poaceascoma taiwanense, Saproamanita manicata, Spegazzinia camelliae, Submersispora variabilis, Thyronectria caudata, T. mackenziei, Tubeufia chiangmaiensis, T. roseohelicospora, Vaginatispora nypae, Wicklowia submersa, Xanthagaricus necopinatus and Xylaria haemorrhoidalis. The data presented herein are based on morphological examination of fresh specimens, coupled with analysis of phylogenetic sequence data to better integrate taxa into appropriate taxonomic ranks and infer their evolutionary relationships.
Keywords: 51 new records; 72 new taxa; Agaricomycetes; Ascomycota; Bartheletiomycetes; Basidiomycota; Dothideomycetes; Eurotiomycetes; Exobasidiomycetes; Laboulbeniomycetes; Leotiomycetes; Mortierellomycetes; Mortierellomycota; Mucoromycetes; Mucoromycota; One new combination; One reference specimen; Phylogeny; Sordariomycetes; Taxonomy.
© MUSHROOM RESEARCH FOUNDATION 2021.
Conflict of interest statement
Conflict of interestThe authors declare that they have no conflict of interest.
Figures





















































































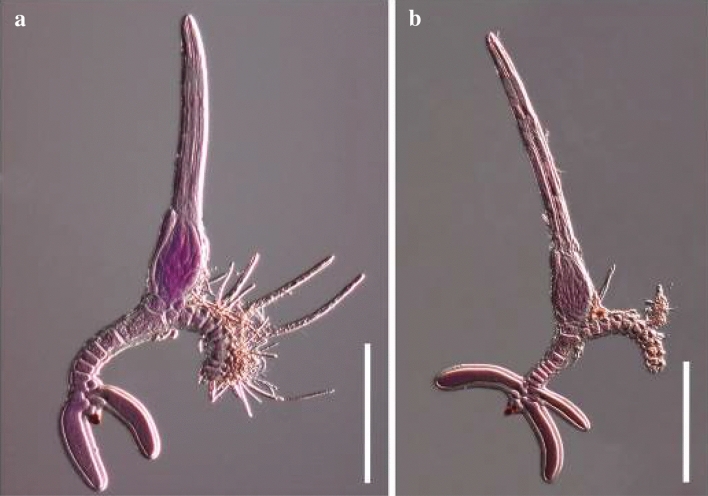

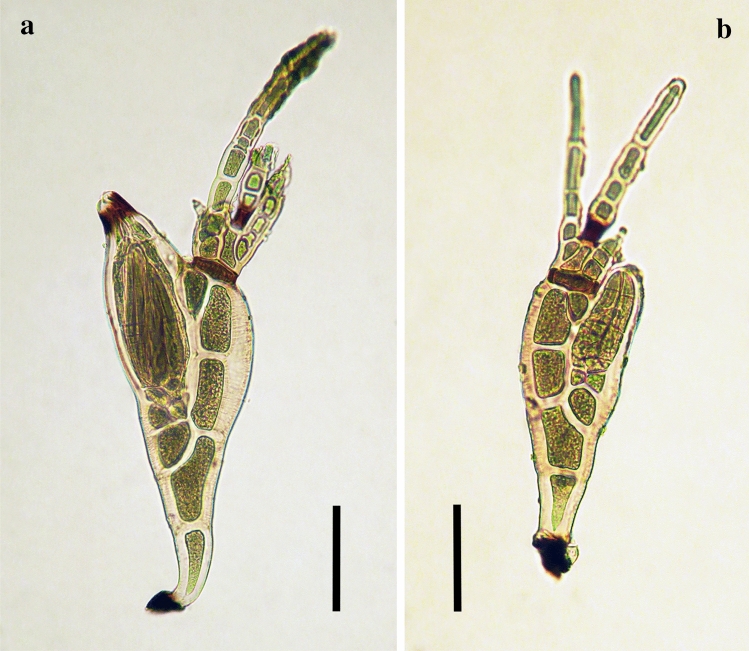





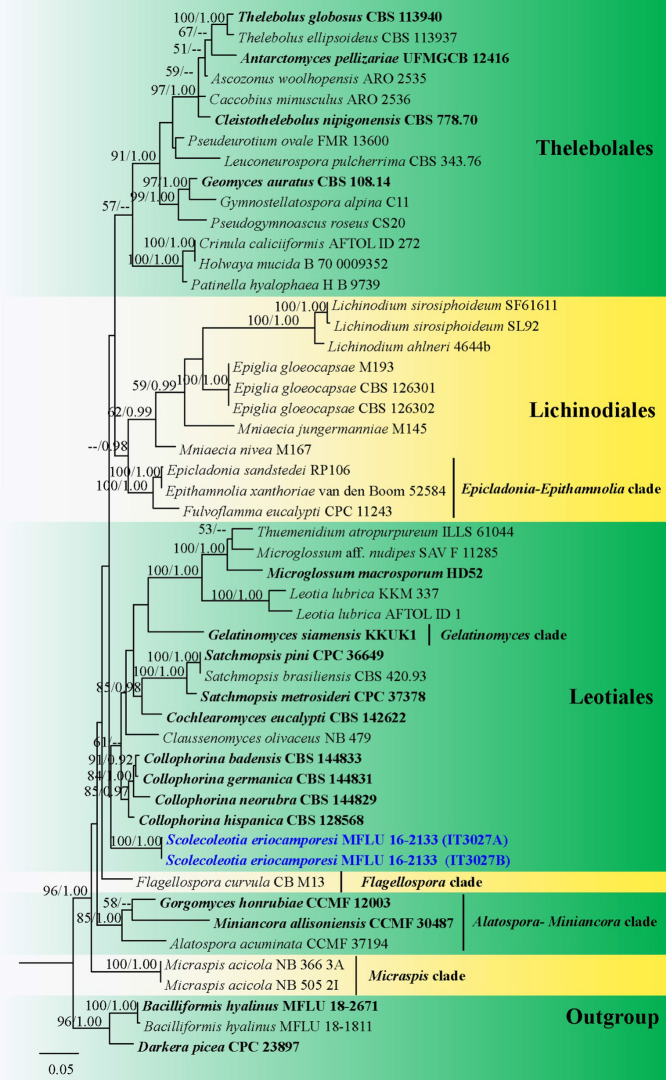






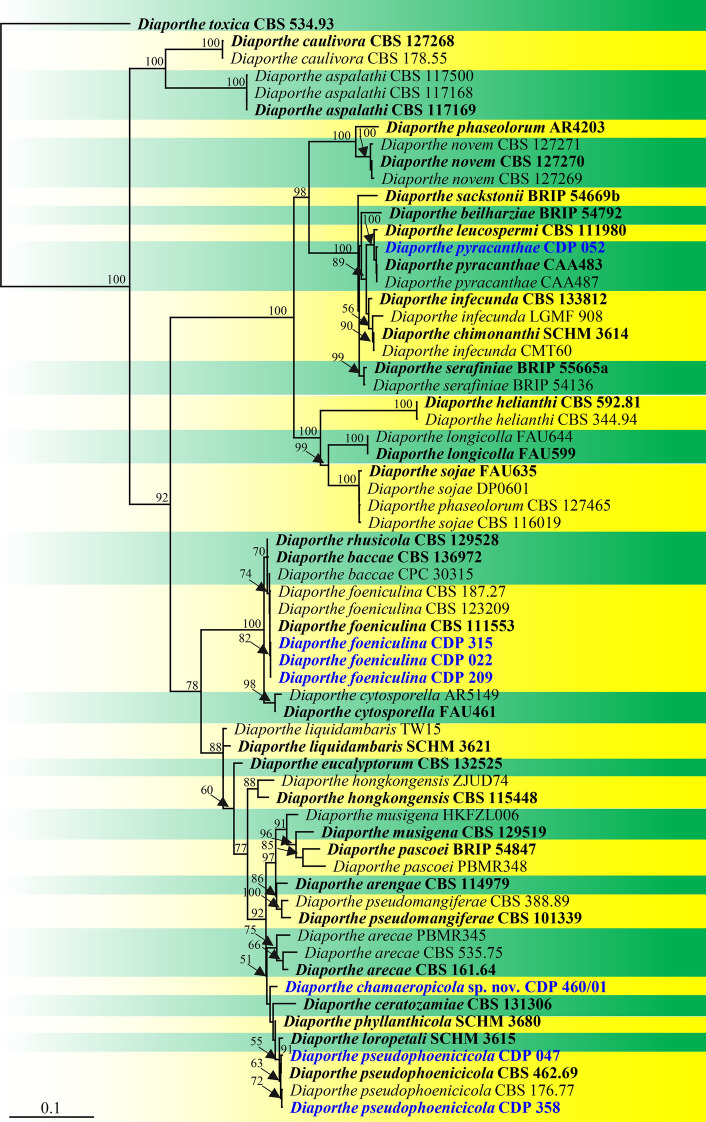



























































































References
-
- Abdel-Wahab MA, El-Sharouny HM, Jones EBG. Two new intertidal lignicolous Swampomyces species from Red Sea mangroves in Egypt. Fungal Divers. 2001;8:35–40.
-
- Abdel-Wahab MA, El-Samawaty AMA, El Gorban AM, Yassin MA, Alsaadi MH. Khaleijomyces marinus gen. et sp. nov. (Juncigenaceae, Torpedosporales) a new lignicolous marine fungus from Saudi Arabia. Phytotaxa. 2018;340:277–285. doi: 10.11646/phytotaxa.340.3.8. - DOI
-
- Abdel-Wahab MA, Jones EBG, Bahkali AHA, El-Gorban AM. Marine fungi from Red Sea mangroves in Saudi Arabia with Fulvocentrum rubrum sp. nov. (Torpedosporales, Ascomycota) Nova Hedwig. 2019;108:365–377. doi: 10.1127/nova_hedwigia/2018/0511. - DOI
-
- Abdollahzadeh J. Diplodia bulgarica, as a new pathogen and potential threat to the apple industry in Iran. Phytopathol Mediterr. 2015;54:128–132.
Grants and funding
LinkOut - more resources
Full Text Sources