

Gaussian accelerated molecular dynamics (GaMD): principles and applications
- PMID: 34899998
- PMCID: PMC8658739
- DOI: 10.1002/wcms.1521
Gaussian accelerated molecular dynamics (GaMD): principles and applications
Abstract
Gaussian accelerated molecular dynamics (GaMD) is a robust computational method for simultaneous unconstrained enhanced sampling and free energy calculations of biomolecules. It works by adding a harmonic boost potential to smooth biomolecular potential energy surface and reduce energy barriers. GaMD greatly accelerates biomolecular simulations by orders of magnitude. Without the need to set predefined reaction coordinates or collective variables, GaMD provides unconstrained enhanced sampling and is advantageous for simulating complex biological processes. The GaMD boost potential exhibits a Gaussian distribution, thereby allowing for energetic reweighting via cumulant expansion to the second order (i.e., "Gaussian approximation"). This leads to accurate reconstruction of free energy landscapes of biomolecules. Hybrid schemes with other enhanced sampling methods, such as the replica exchange GaMD (rex-GaMD) and replica exchange umbrella sampling GaMD (GaREUS), have also been introduced, further improving sampling and free energy calculations. Recently, new "selective GaMD" algorithms including the ligand GaMD (LiGaMD) and peptide GaMD (Pep-GaMD) enabled microsecond simulations to capture repetitive dissociation and binding of small-molecule ligands and highly flexible peptides. The simulations then allowed highly efficient quantitative characterization of the ligand/peptide binding thermodynamics and kinetics. Taken together, GaMD and its innovative variants are applicable to simulate a wide variety of biomolecular dynamics, including protein folding, conformational changes and allostery, ligand binding, peptide binding, protein-protein/nucleic acid/carbohydrate interactions, and carbohydrate/nucleic acid interactions. In this review, we present principles of the GaMD algorithms and recent applications in biomolecular simulations and drug design.
Conflict of interest statement
Conflict of Interest The authors have declared no conflict of interest for this article.
Figures
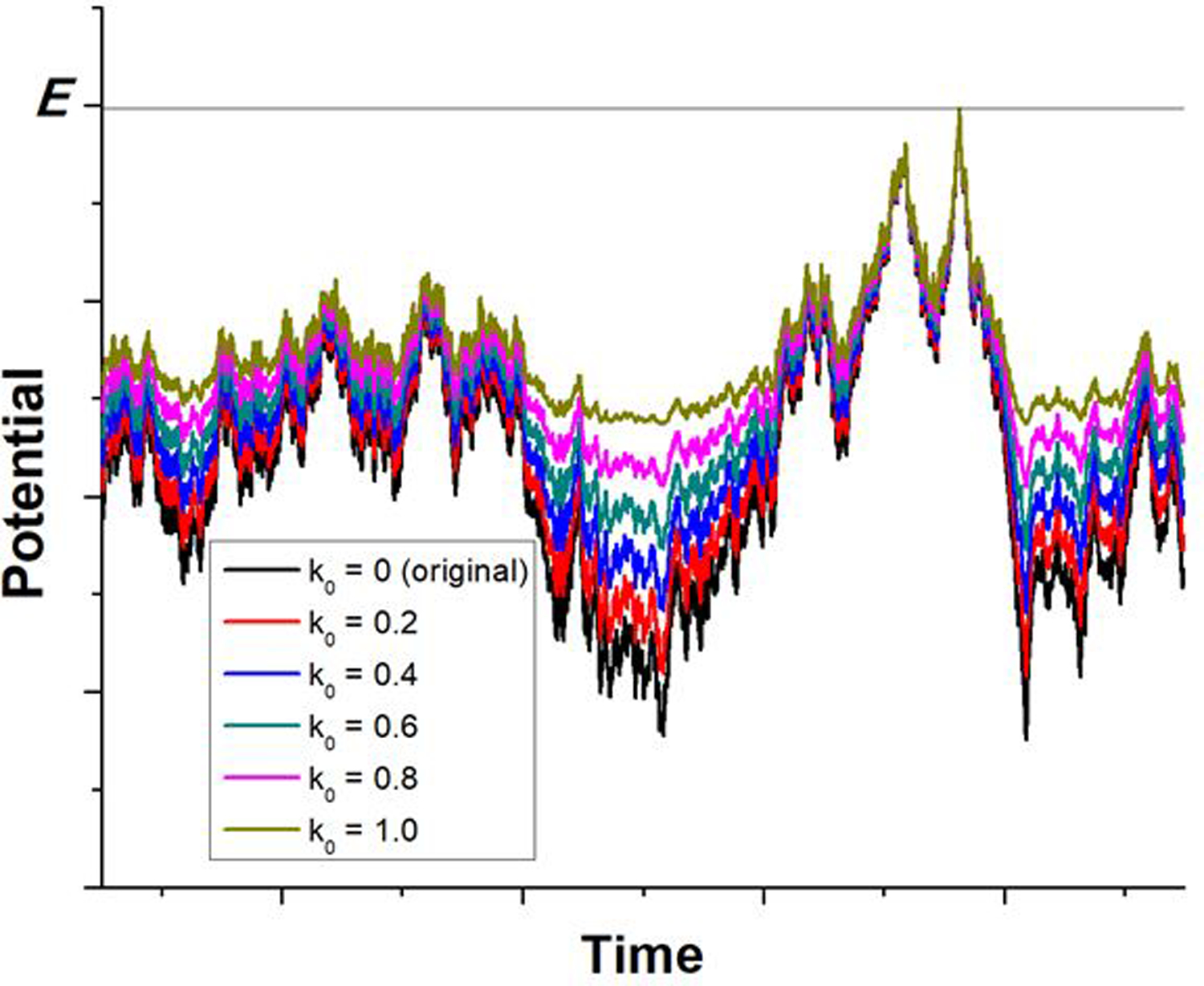

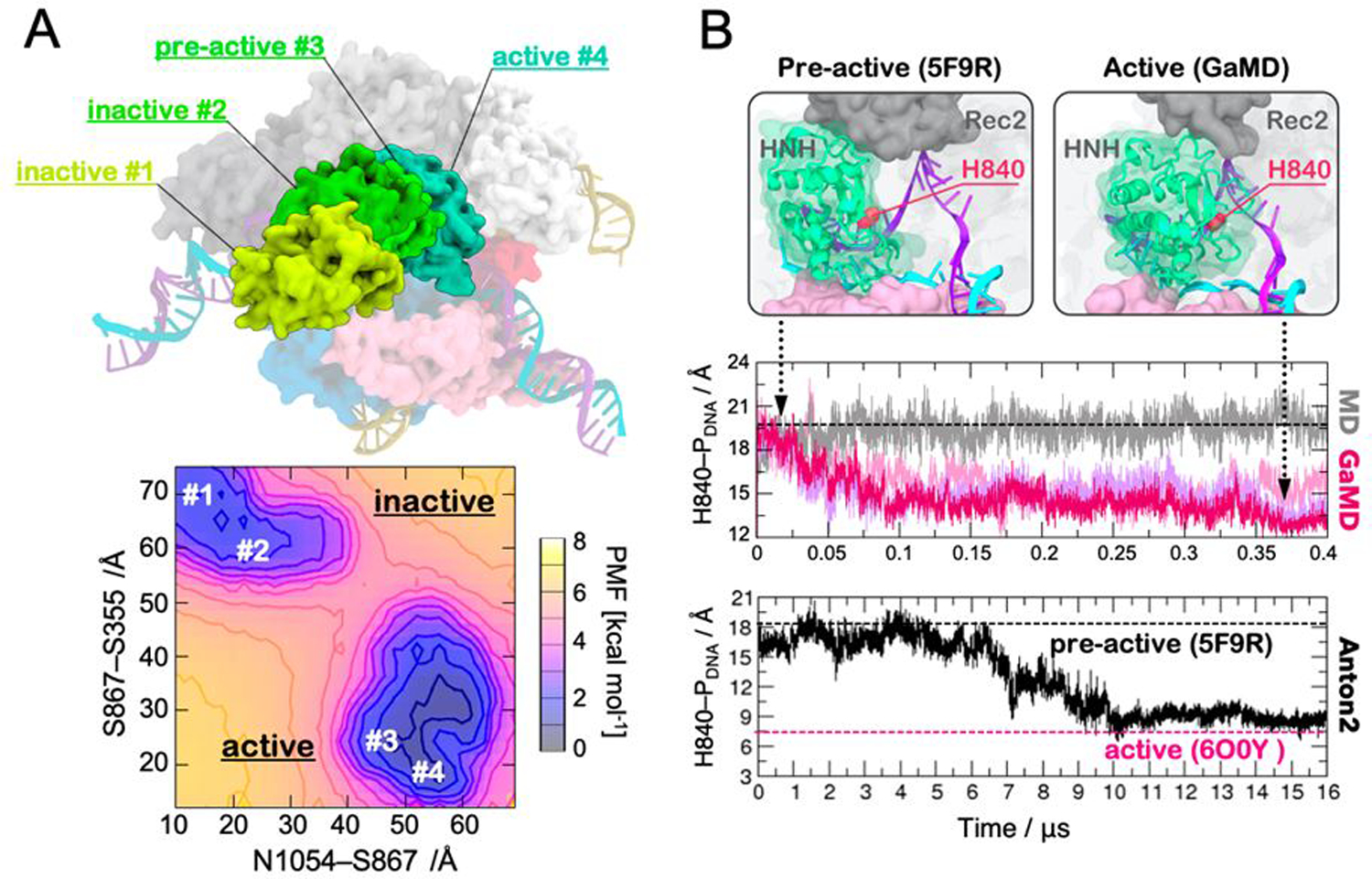


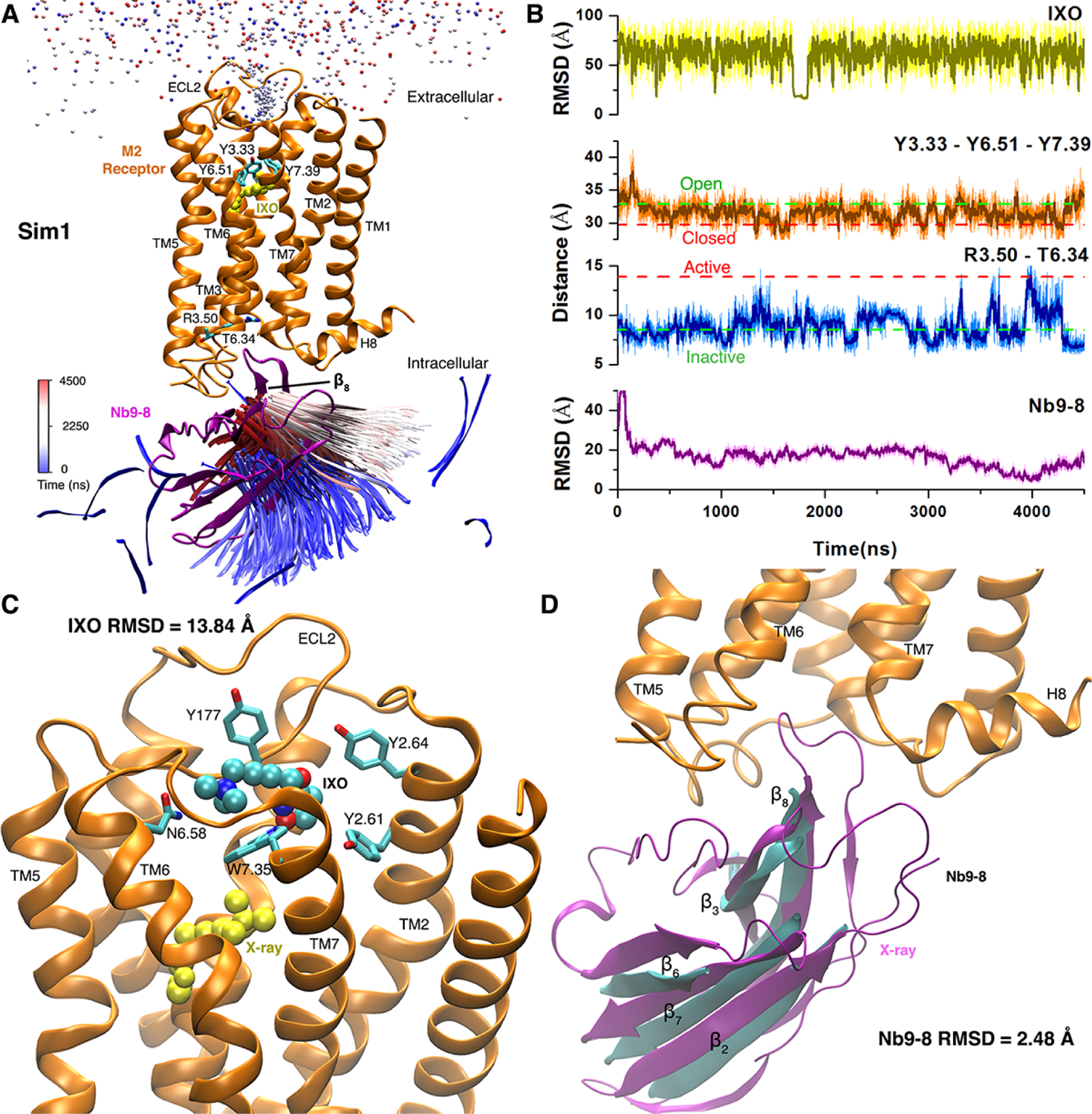
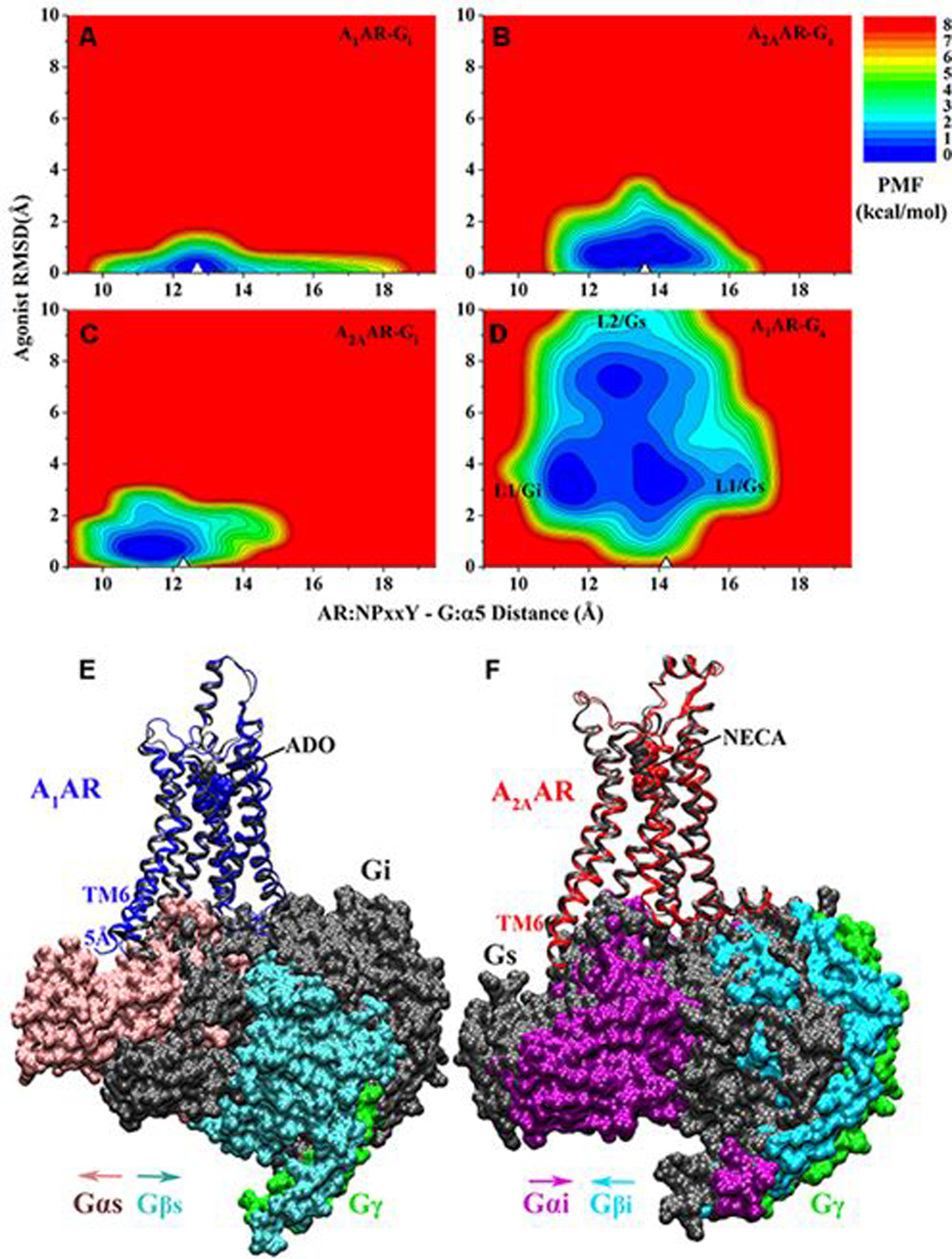

References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous