

Targeting RAS phosphorylation in cancer therapy: Mechanisms and modulators
- PMID: 34900528
- PMCID: PMC8642438
- DOI: 10.1016/j.apsb.2021.02.014
Targeting RAS phosphorylation in cancer therapy: Mechanisms and modulators
Abstract
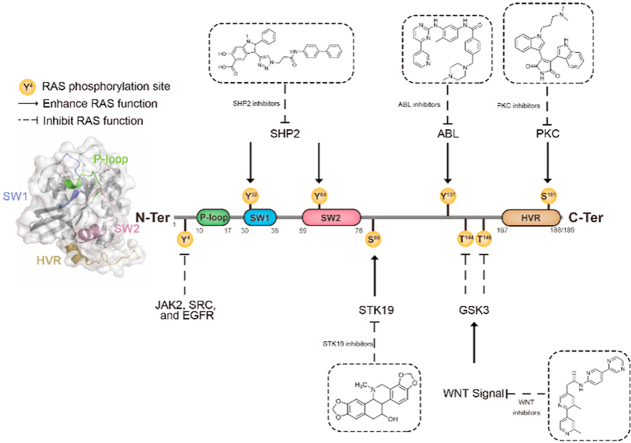
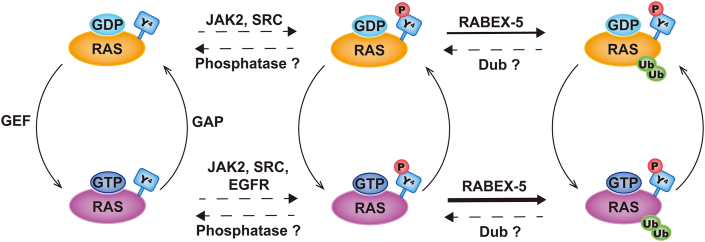
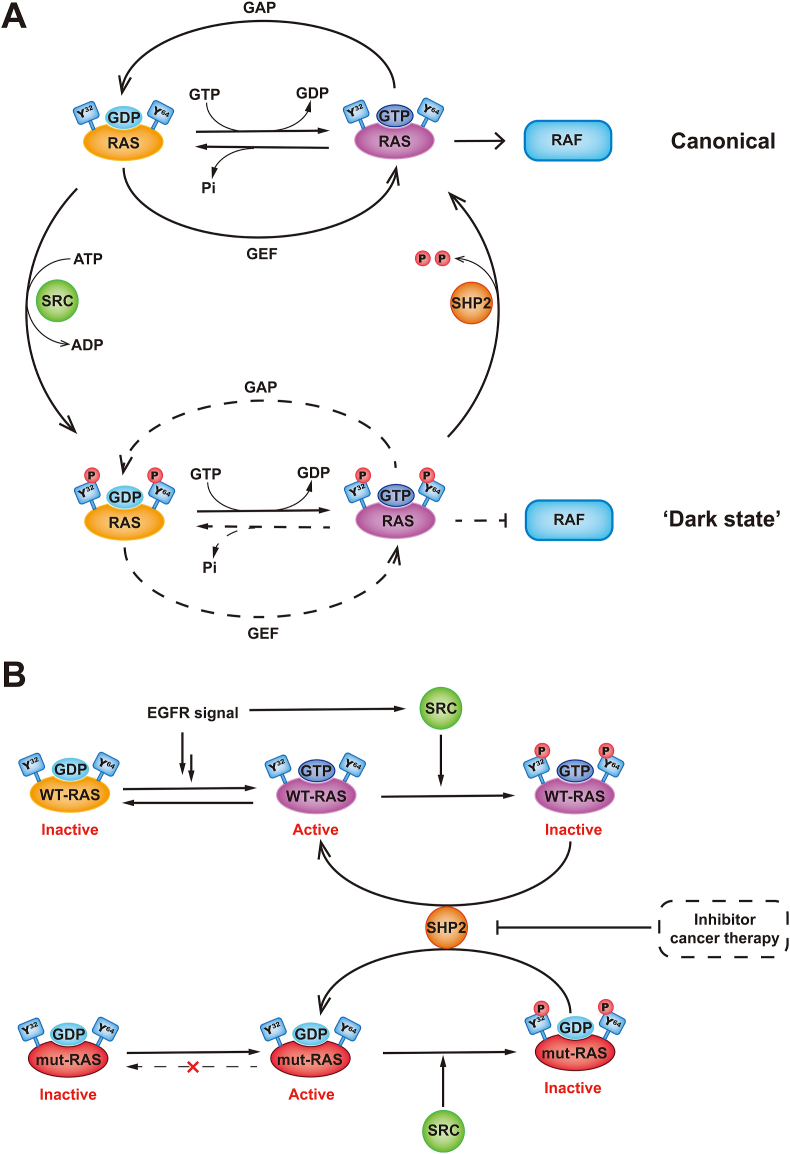
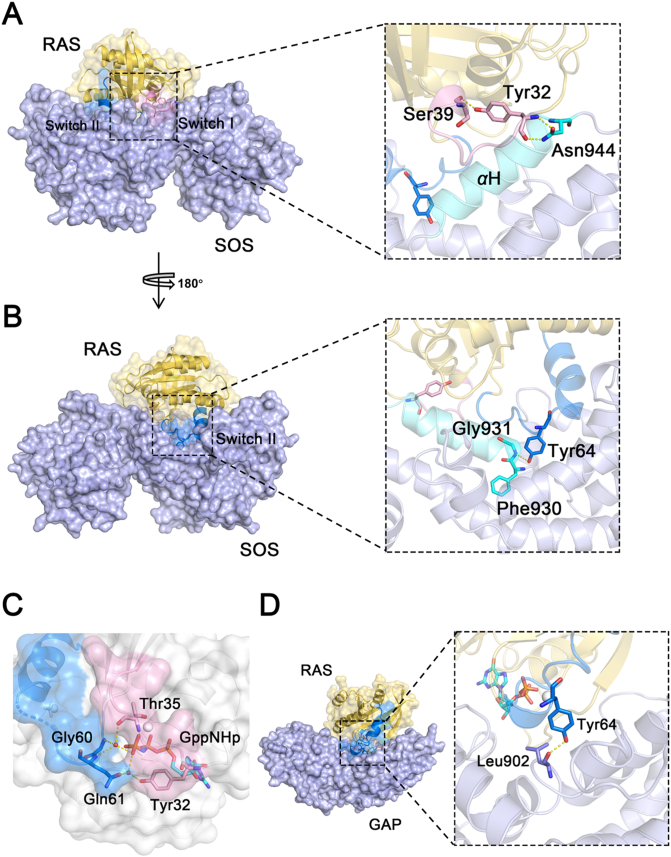
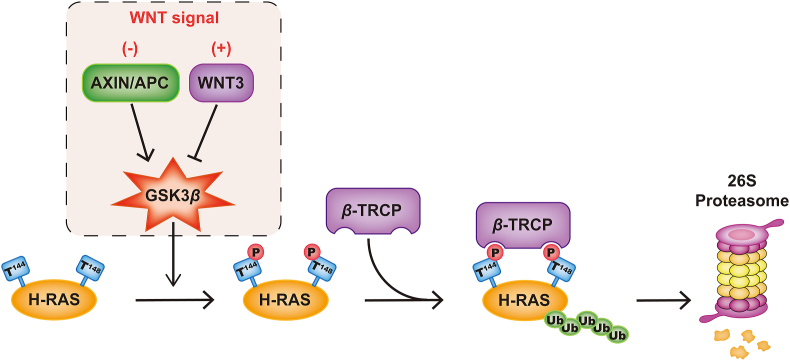
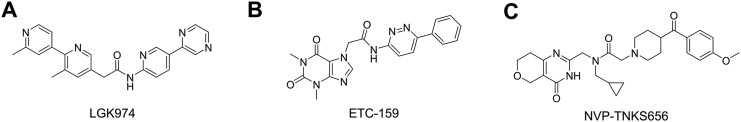
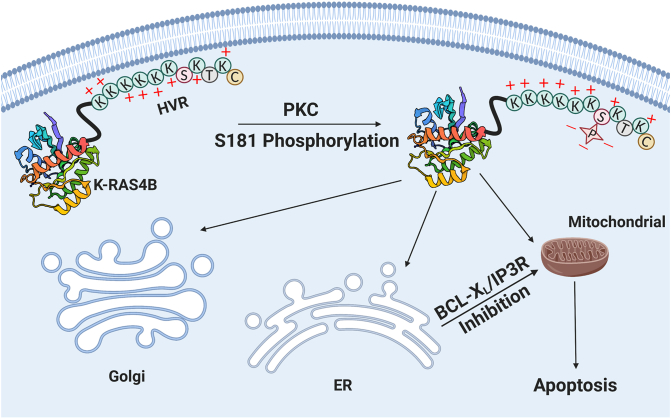
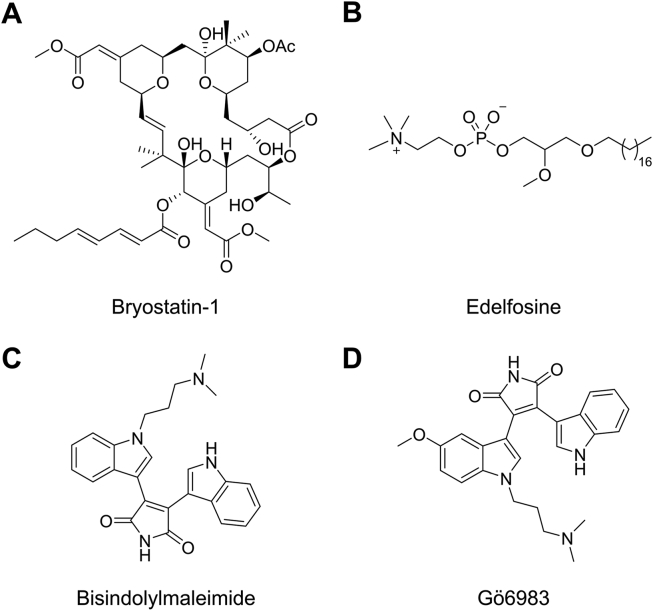
RAS, a member of the small GTPase family, functions as a binary switch by shifting between inactive GDP-loaded and active GTP-loaded state. RAS gain-of-function mutations are one of the leading causes in human oncogenesis, accounting for ∼19% of the global cancer burden. As a well-recognized target in malignancy, RAS has been intensively studied in the past decades. Despite the sustained efforts, many failures occurred in the earlier exploration and resulted in an 'undruggable' feature of RAS proteins. Phosphorylation at several residues has been recently determined as regulators for wild-type and mutated RAS proteins. Therefore, the development of RAS inhibitors directly targeting the RAS mutants or towards upstream regulatory kinases supplies a novel direction for tackling the anti-RAS difficulties. A better understanding of RAS phosphorylation can contribute to future therapeutic strategies. In this review, we comprehensively summarized the current advances in RAS phosphorylation and provided mechanistic insights into the signaling transduction of associated pathways. Importantly, the preclinical and clinical success in developing anti-RAS drugs targeting the upstream kinases and potential directions of harnessing allostery to target RAS phosphorylation sites were also discussed.
Keywords: ABL, Abelson; APC, adenomatous polyposis coli; Allostery; CK1, casein kinase 1; CML, chronic myeloid leukemia; ER, endoplasmic reticulum; GAPs, GTPase-activating proteins; GEFs, guanine nucleotide exchange-factors; GSK3, glycogen synthase kinase 3; HVR, hypervariable region; IP3R, inositol trisphosphate receptors; LRP6, lipoprotein-receptor-related protein 6; OMM, outer mitochondrial membrane; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PPIs, protein−protein interactions; Phosphorylation; Protein kinases; RAS; RIN1, RAB-interacting protein 1; SHP2, SRC homology 2 domain containing phosphatase 2; SOS, Son of Sevenless; STK19, serine/threonine-protein kinase 19; TKIs, tyrosine kinase inhibitors; Undruggable.
© 2021 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Figures
Similar articles
-
Localizing a control region in the pathway to leukotriene C(4) secretion following stimulation of human basophils with anti-IgE antibody.J Immunol. 2001 Dec 15;167(12):7027-37. doi: 10.4049/jimmunol.167.12.7027. J Immunol. 2001. PMID: 11739523
-
Mechanism of SHIP-mediated inhibition of insulin- and platelet-derived growth factor-stimulated mitogen-activated protein kinase activity in 3T3-L1 adipocytes.Mol Endocrinol. 2005 Feb;19(2):421-30. doi: 10.1210/me.2004-0096. Epub 2004 Oct 14. Mol Endocrinol. 2005. PMID: 15486046
-
Ras activation in response to phorbol ester proceeds independently of the EGFR via an unconventional nucleotide-exchange factor system in COS-7 cells.Biochem J. 2006 Sep 1;398(2):243-56. doi: 10.1042/BJ20060160. Biochem J. 2006. PMID: 16709153 Free PMC article.
-
Activation of Ras by receptor tyrosine kinases.J Am Soc Nephrol. 1994 Dec;5(6):1288-99. doi: 10.1681/ASN.V561288. J Am Soc Nephrol. 1994. PMID: 7893993 Review.
-
Rit subfamily small GTPases: regulators in neuronal differentiation and survival.Cell Signal. 2013 Oct;25(10):2060-8. doi: 10.1016/j.cellsig.2013.06.002. Epub 2013 Jun 11. Cell Signal. 2013. PMID: 23770287 Free PMC article. Review.
Cited by
-
Discovery of Hit Compounds Targeting the P4 Allosteric Site of K-RAS, Identified through Ensemble-Based Virtual Screening.J Chem Inf Model. 2023 Oct 23;63(20):6412-6422. doi: 10.1021/acs.jcim.3c01212. Epub 2023 Oct 12. J Chem Inf Model. 2023. PMID: 37824186 Free PMC article.
-
In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy.Int J Mol Sci. 2025 Apr 2;26(7):3293. doi: 10.3390/ijms26073293. Int J Mol Sci. 2025. PMID: 40244134 Free PMC article.
-
Pan-KRAS inhibitors suppress proliferation through feedback regulation in pancreatic ductal adenocarcinoma.Acta Pharmacol Sin. 2022 Oct;43(10):2696-2708. doi: 10.1038/s41401-022-00897-4. Epub 2022 Mar 29. Acta Pharmacol Sin. 2022. PMID: 35352018 Free PMC article.
-
Elevated Levels of Mislocalised, Constitutive Ras Signalling Can Drive Quiescence by Uncoupling Cell-Cycle Regulation from Metabolic Homeostasis.Biomolecules. 2023 Nov 6;13(11):1619. doi: 10.3390/biom13111619. Biomolecules. 2023. PMID: 38002301 Free PMC article.
-
Mechanistic Insights Into Co-Administration of Allosteric and Orthosteric Drugs to Overcome Drug-Resistance in T315I BCR-ABL1.Front Pharmacol. 2022 Mar 18;13:862504. doi: 10.3389/fphar.2022.862504. eCollection 2022. Front Pharmacol. 2022. PMID: 35370687 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
