

HDAC2 Facilitates Pancreatic Cancer Metastasis
- PMID: 34903606
- PMCID: PMC9359718
- DOI: 10.1158/0008-5472.CAN-20-3209
HDAC2 Facilitates Pancreatic Cancer Metastasis
Abstract
The mortality of patients with pancreatic ductal adenocarcinoma (PDAC) is strongly associated with metastasis, a multistep process that is incompletely understood in this disease. Although genetic drivers of PDAC metastasis have not been defined, transcriptional and epigenetic rewiring can contribute to the metastatic process. The epigenetic eraser histone deacetylase 2 (HDAC2) has been connected to less differentiated PDAC, but the function of HDAC2 in PDAC has not been comprehensively evaluated. Using genetically defined models, we show that HDAC2 is a cellular fitness factor that controls cell cycle in vitro and metastasis in vivo, particularly in undifferentiated, mesenchymal PDAC cells. Unbiased expression profiling detected a core set of HDAC2-regulated genes. HDAC2 controlled expression of several prosurvival receptor tyrosine kinases connected to mesenchymal PDAC, including PDGFRα, PDGFRβ, and EGFR. The HDAC2-maintained program disabled the tumor-suppressive arm of the TGFβ pathway, explaining impaired metastasis formation of HDAC2-deficient PDAC. These data identify HDAC2 as a tractable player in the PDAC metastatic cascade. The complexity of the function of epigenetic regulators like HDAC2 implicates that an increased understanding of these proteins is needed for implementation of effective epigenetic therapies.
Significance: HDAC2 has a context-specific role in undifferentiated PDAC and the capacity to disseminate systemically, implicating HDAC2 as targetable protein to prevent metastasis.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
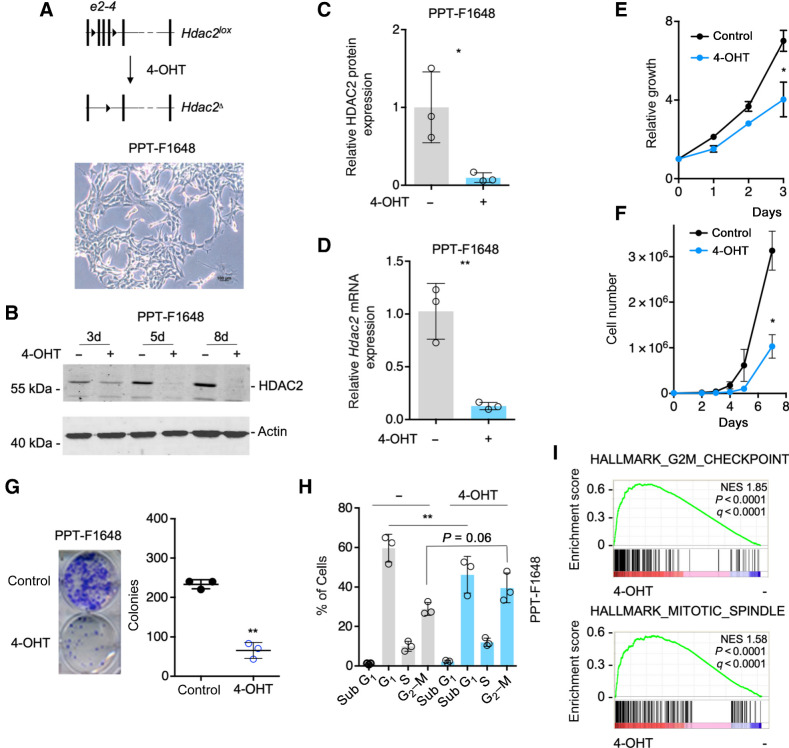
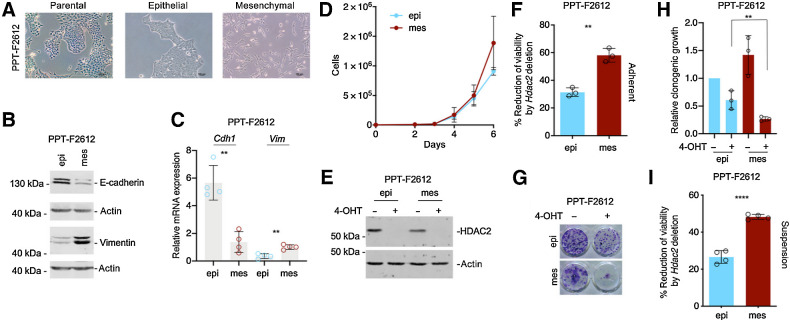
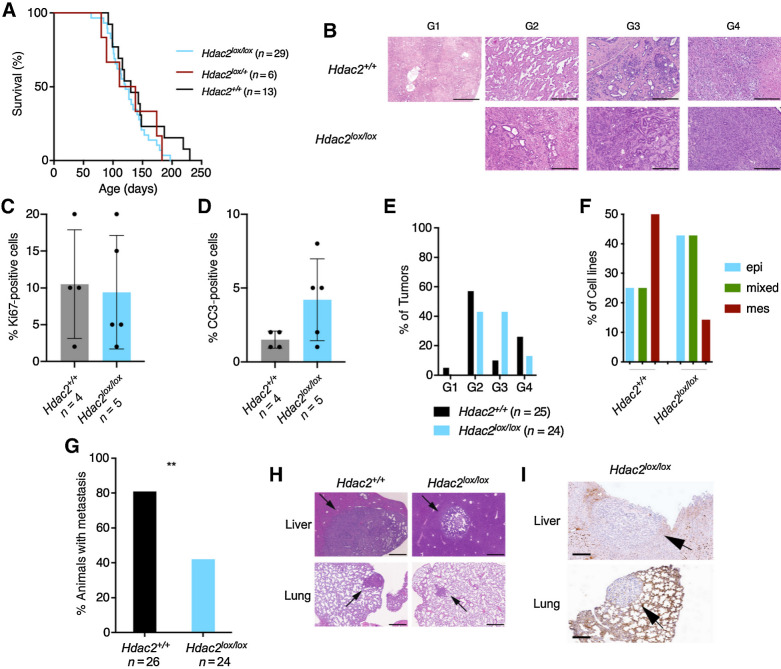
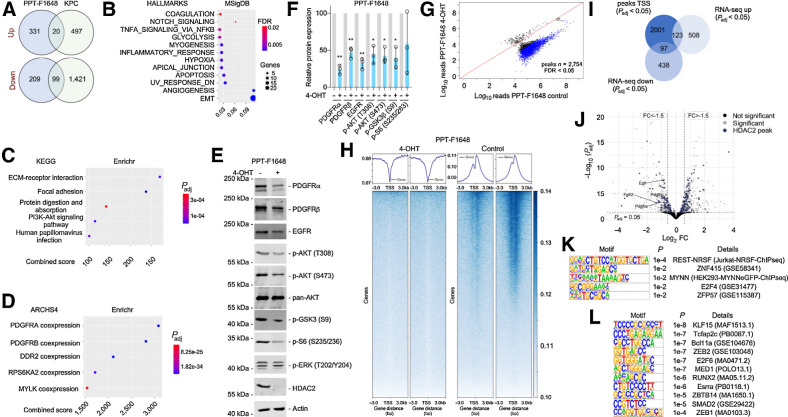
References
-
- Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O'Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020;52:231–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
