

Targeting Chemotherapy to Decondensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression
- PMID: 34903608
- PMCID: PMC8889548
- DOI: 10.1158/0008-5472.CAN-21-1297
Targeting Chemotherapy to Decondensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression
Abstract
Despite treatment with intensive chemotherapy, acute myelogenous leukemia (AML) remains an aggressive malignancy with a dismal outcome in most patients. We found that AML cells exhibit an unusually rapid accumulation of the repressive histone mark H3K27me3 on nascent DNA. In cell lines, primary cells and xenograft mouse models, inhibition of the H3K27 histone methyltransferase EZH2 to decondense the H3K27me3-marked chromatin of AML cells enhanced chromatin accessibility and chemotherapy-induced DNA damage, apoptosis, and leukemia suppression. These effects were further promoted when chromatin decondensation of AML cells was induced upon S-phase entry after release from a transient G1 arrest mediated by CDK4/6 inhibition. In the p53-null KG-1 and THP-1 AML cell lines, EZH2 inhibitor and doxorubicin cotreatment induced transcriptional reprogramming that was, in part, dependent on derepression of H3K27me3-marked gene promoters and led to increased expression of cell death-promoting and growth-inhibitory genes.In conclusion, decondensing H3K27me3-marked chromatin by EZH2 inhibition represents a promising approach to improve the efficacy of DNA-damaging cytotoxic agents in patients with AML. This strategy might allow for a lowering of chemotherapy doses, with a consequent reduction of treatment-related side effects in elderly patients with AML or those with significant comorbidities. SIGNIFICANCE: Pharmacological inhibition of EZH2 renders DNA of AML cells more accessible to cytotoxic agents, facilitating leukemia suppression with reduced doses of chemotherapy.See related commentary by Adema and Colla, p. 359.
©2021 American Association for Cancer Research.
Figures
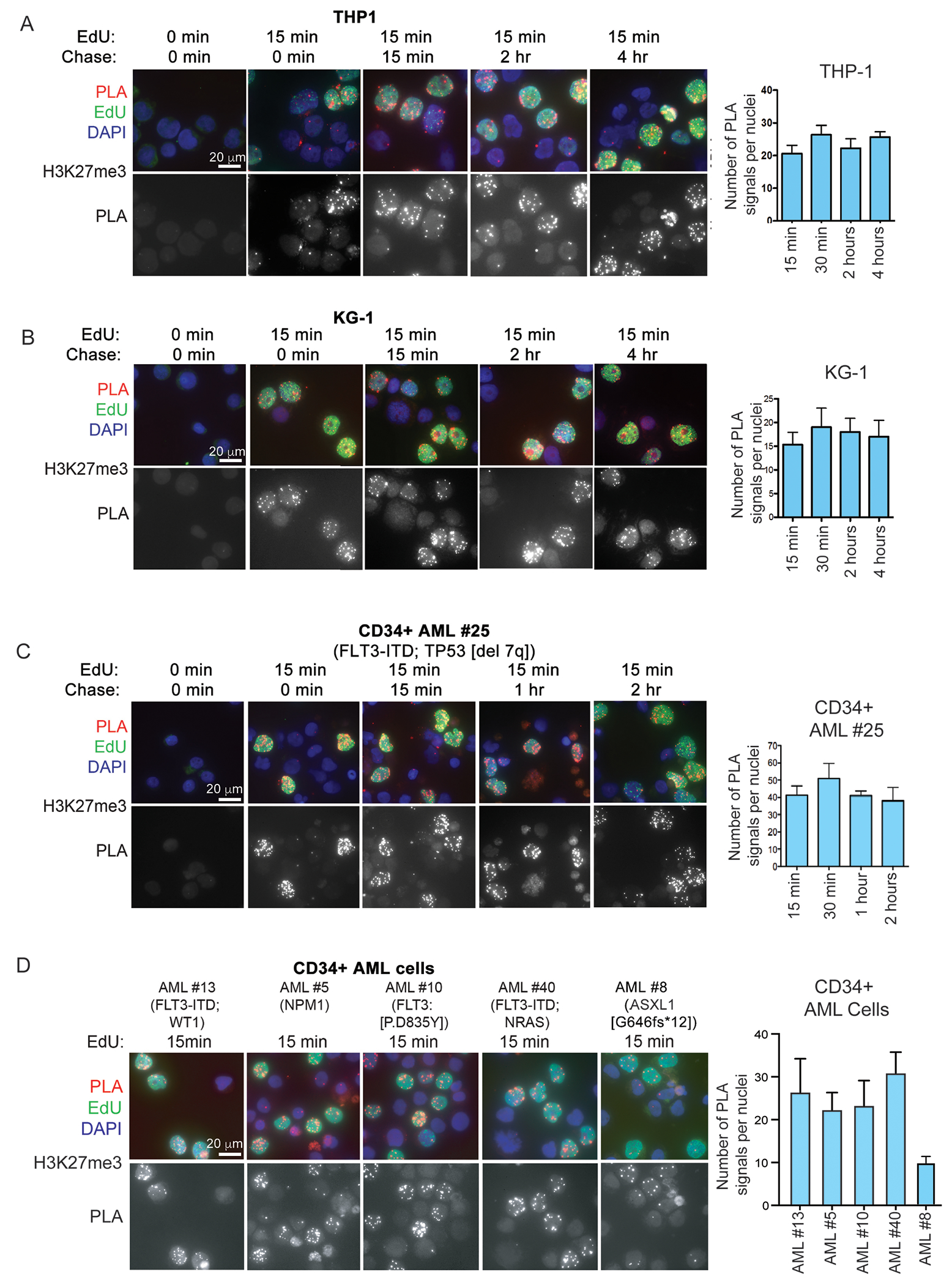
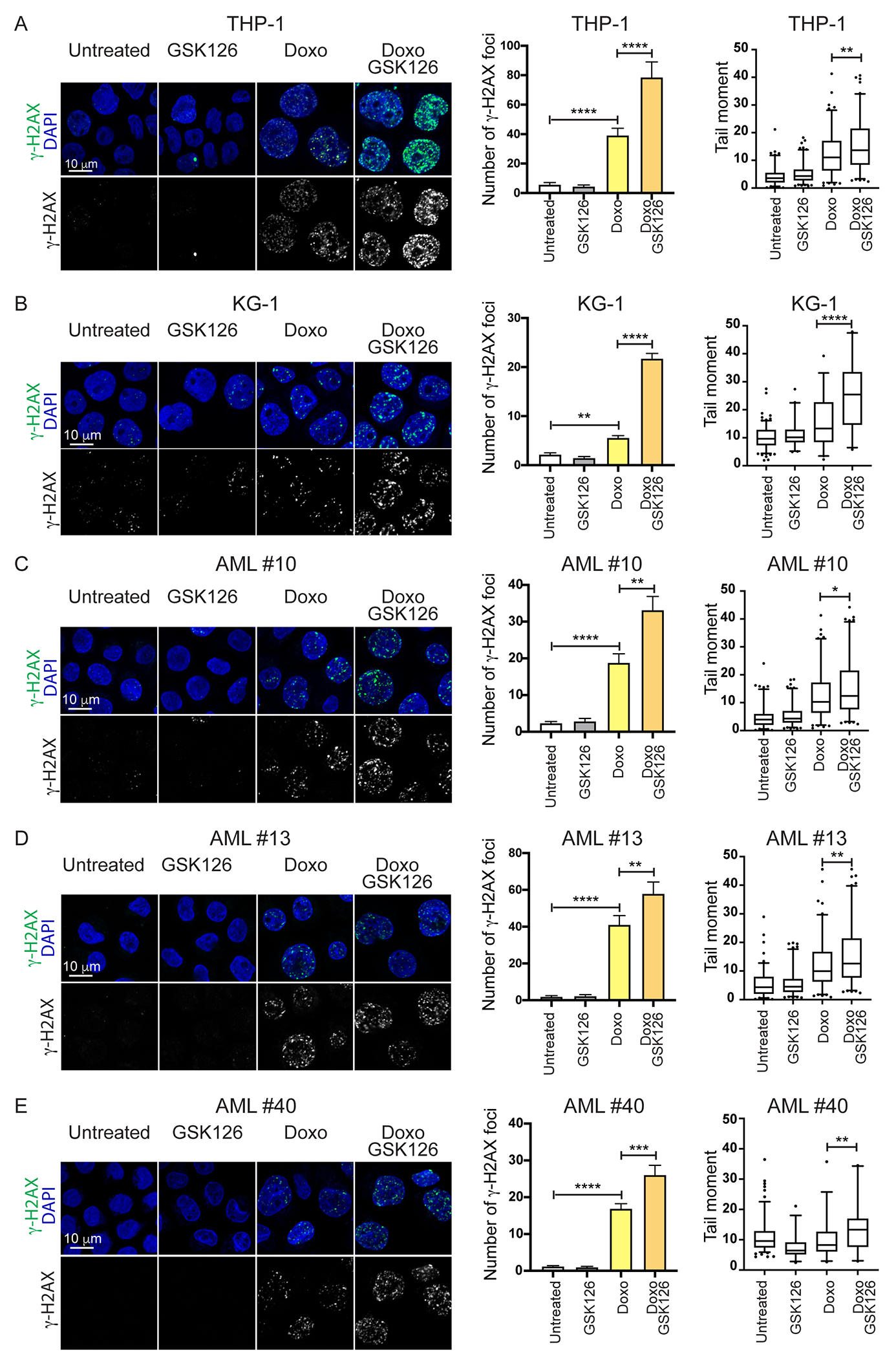
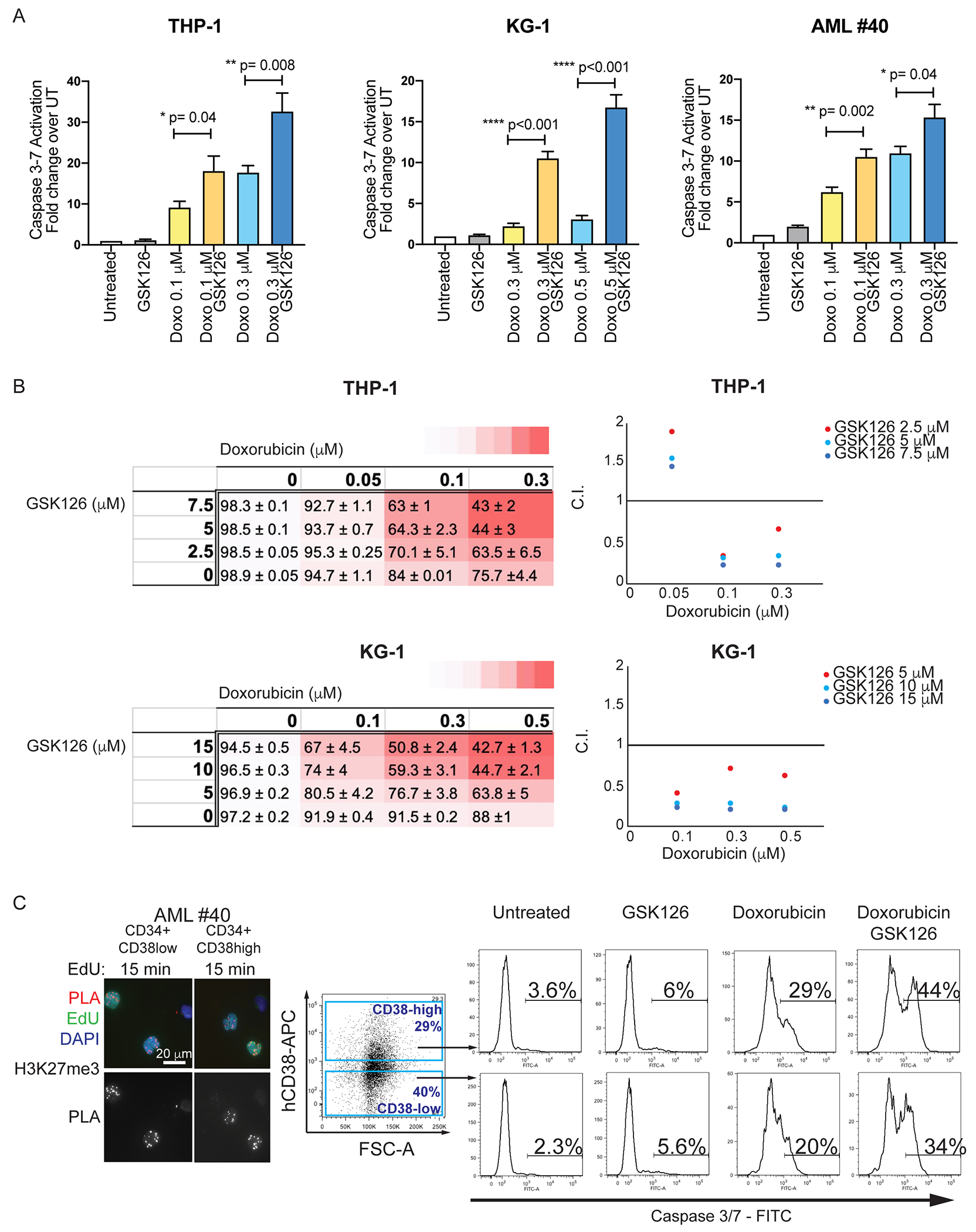
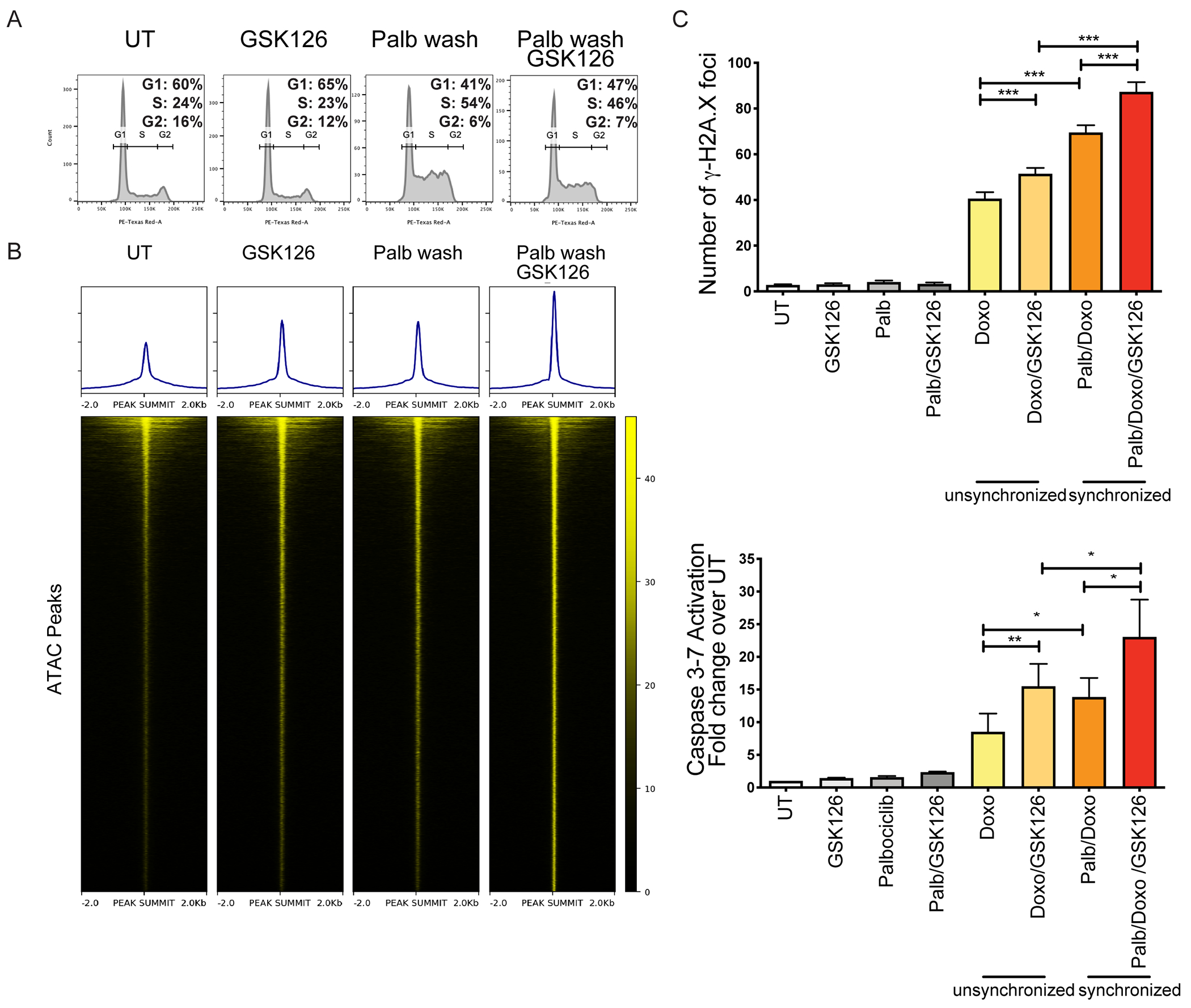
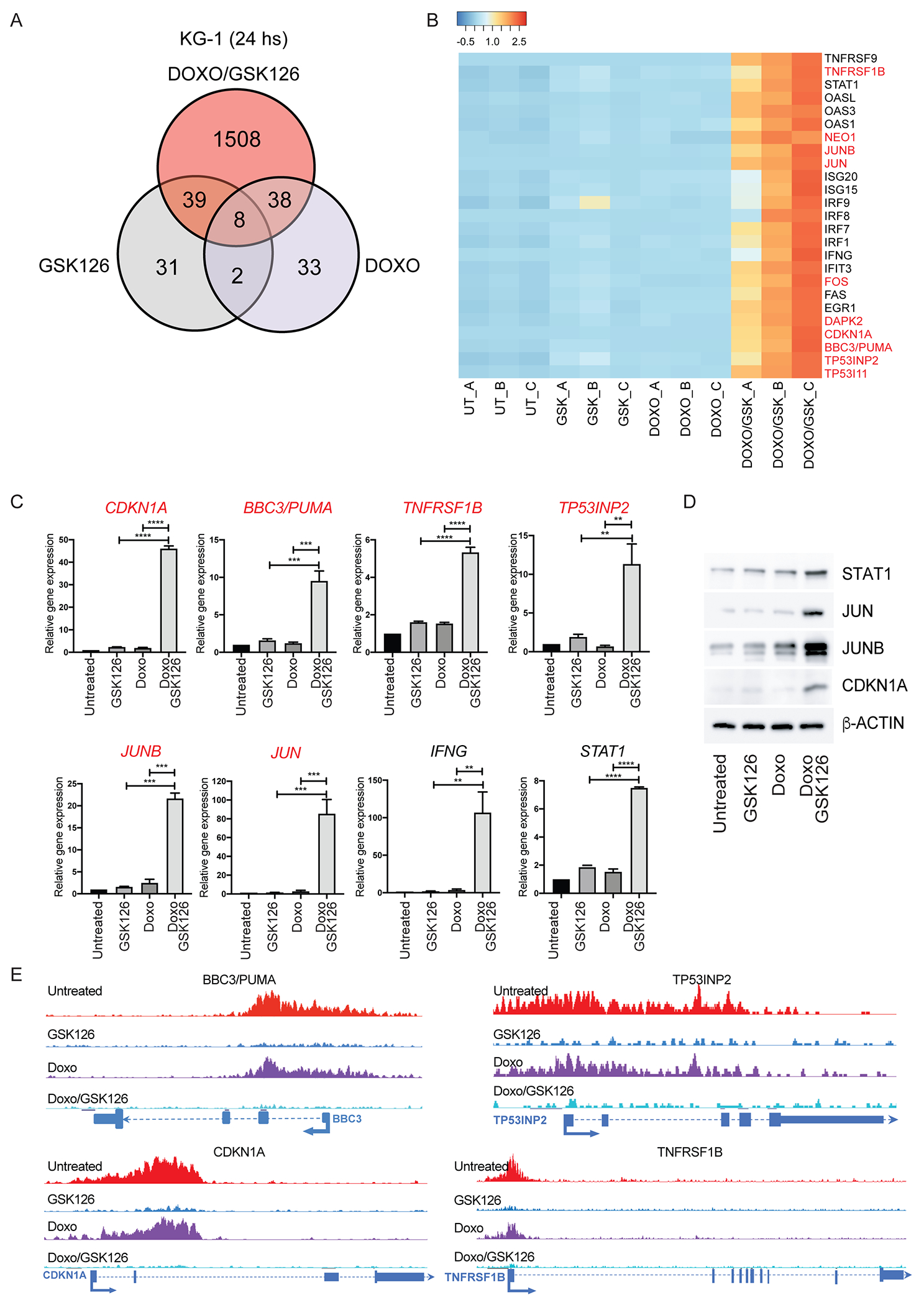
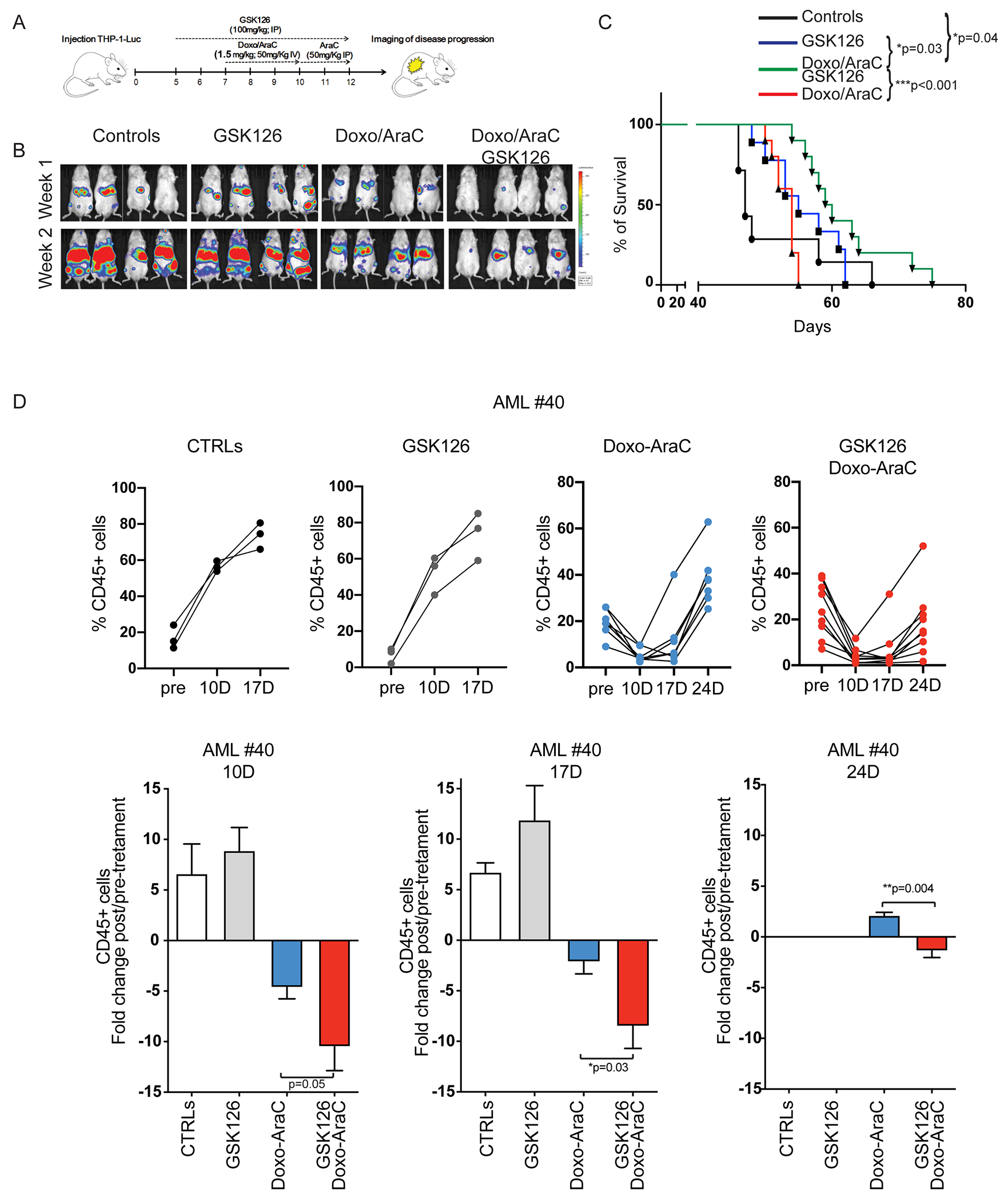
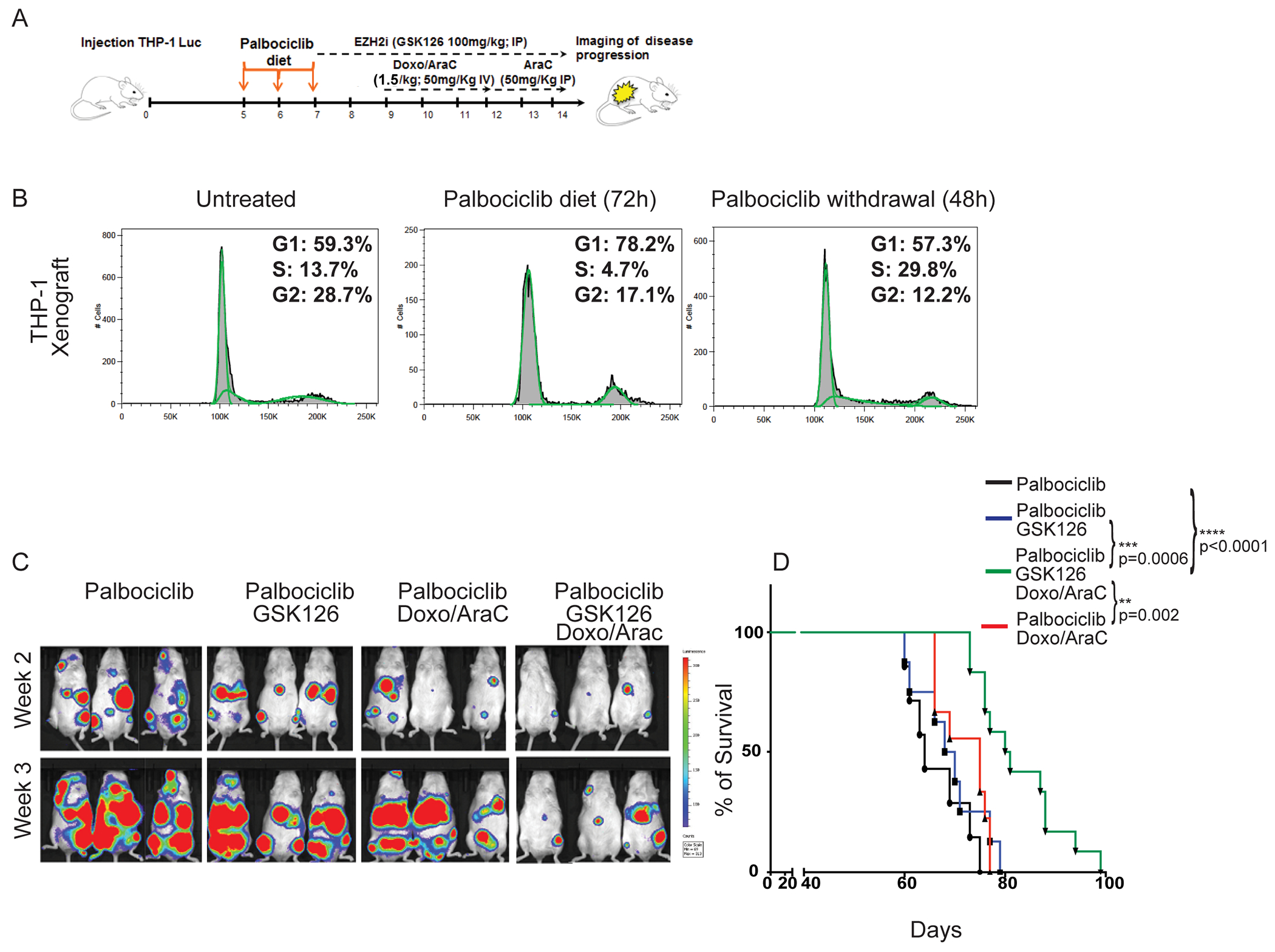
Comment in
-
EZH2 Inhibitors: The Unpacking Revolution.Cancer Res. 2022 Feb 1;82(3):359-361. doi: 10.1158/0008-5472.CAN-21-4311. Cancer Res. 2022. PMID: 35110396
References
-
- Dohner H, Weisdorf DJ, and Bloomfield C. Acute myeloid leukemia. N Engl J Med 2015; 373:1136–1152. - PubMed
-
- Papaemmanuil E, Gerstung M, Bullinger L, Potter NE, Henser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, Ganly P, Mudie L, McLaren S, O’Meara S, Reine K, Jones DR, Teague JW, Butler AP, Greaves MF, Ganser A, Dohner K, Schlenk RF, Dohner H, and Campbell PJ. Genome classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209–2221. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
