

Safety and Efficacy of Acetyl-DL-Leucine in Certain Types of Cerebellar Ataxia: The ALCAT Randomized Clinical Crossover Trial
- PMID: 34905009
- PMCID: PMC8672236
- DOI: 10.1001/jamanetworkopen.2021.35841
Safety and Efficacy of Acetyl-DL-Leucine in Certain Types of Cerebellar Ataxia: The ALCAT Randomized Clinical Crossover Trial
Abstract
Importance: Cerebellar ataxia is a neurodegenerative disease impairing motor function characterized by ataxia of stance, gait, speech, and fine motor disturbances.
Objective: To investigate the efficacy, safety, and tolerability of the modified essential amino acid acetyl-DL-leucine in treating patients who have cerebellar ataxia.
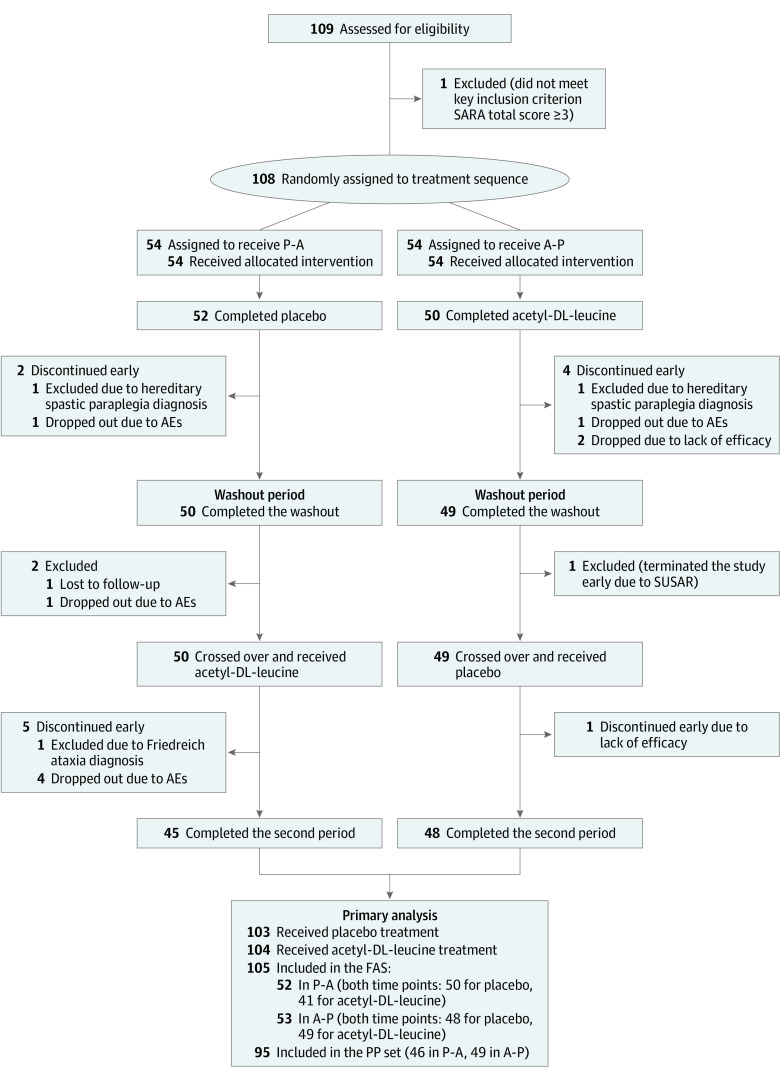
Design, setting, and participants: The Acetyl-DL-leucine on Cerebellar Ataxia (ALCAT) trial was an investigator-initiated, multicenter, double-blind, randomized, placebo-controlled, clinical crossover trial. The study was conducted at 7 university hospitals in Germany and Austria between January 25, 2016, and February 17, 2017. Patients were aged at least 18 years and diagnosed with cerebellar ataxia of hereditary (suspected or genetically confirmed) or nonhereditary or unknown type presenting with a total score of at least 3 points on the Scale for the Assessment and Rating of Ataxia (SARA). Statistical analysis was performed from April 2018 to June 2018 and January 2020 to March 2020.
Interventions: Patients were randomly assigned (1:1) to receive acetyl-DL-leucine orally (5 g per day after 2 weeks up-titration) followed by a matched placebo, each for 6 weeks, separated by a 4-week washout, or vice versa. The randomization was done via a web-based, permuted block-wise randomization list (block size, 2) that was stratified by disease subtype (hereditary vs nonhereditary or unknown) and site.
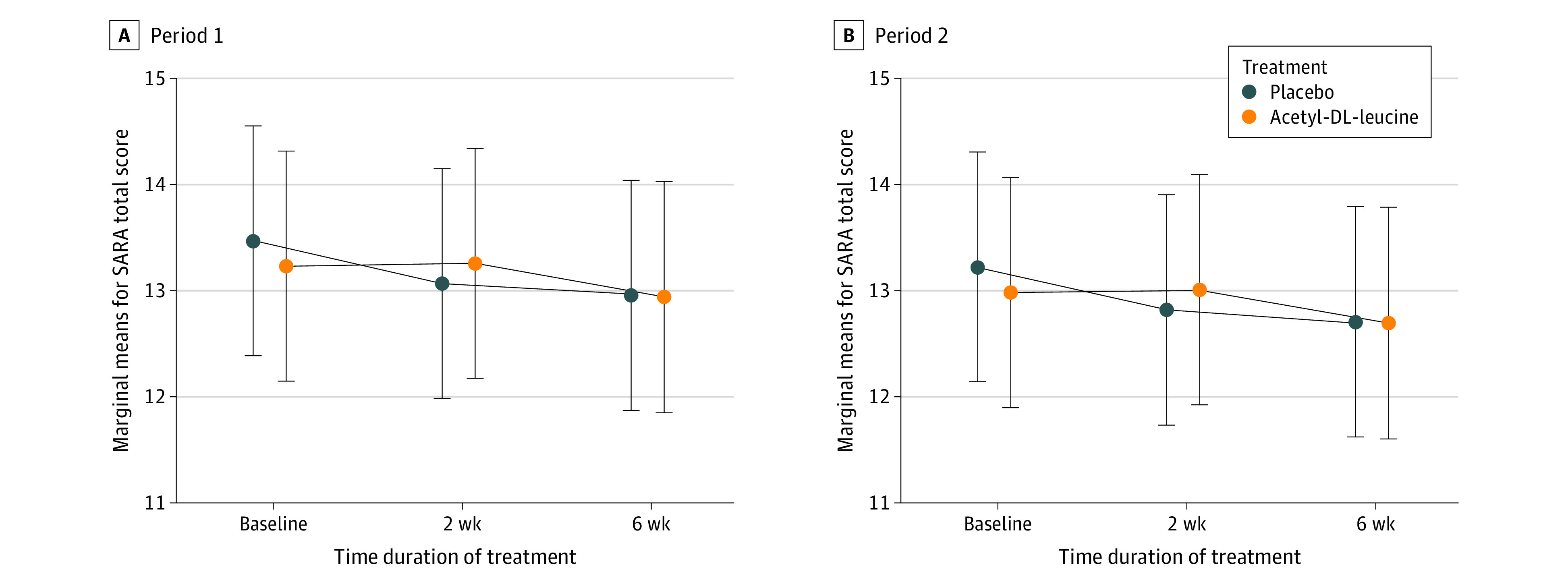
Main outcomes and measures: Primary efficacy outcome was the absolute change of SARA total score from (period-dependent) baseline to week 6.
Results: Among 108 patients who were randomly assigned to sequence groups (54 patients each), 55 (50.9%) were female; the mean (SD) age was 54.8 (14.4) years; and the mean (SD) SARA total score was 13.33 (5.57) points. The full analysis set included 105 patients (80 patients with hereditary, 25 with nonhereditary or unknown cerebellar ataxia). There was no evidence of a difference in the mean absolute change from baseline to week 6 in SARA total scores between both treatments (mean treatment difference: 0.23 points [95% CI, -0.40 to 0.85 points]).
Conclusions and relevance: In this large multicenter, double-blind, randomized, placebo-controlled clinical crossover trial, acetyl-DL-leucine in the investigated dosage and treatment duration was not superior to placebo for the symptomatic treatment of certain types of ataxia. The drug was well tolerated; and ALCAT yielded valuable information about the duration of treatment periods and the role of placebo response in cerebellar ataxia. These findings suggest that further symptom-oriented trials are needed for evaluating the long-term effects of acetyl-DL-leucine for well-defined subgroups of cerebellar ataxia.
Trial registration: EudraCT 2015-000460-34.
Conflict of interest statement
Figures
Comment in
-
Clinical trials for cerebellar ataxia.J Neurol. 2022 May;269(5):2819-2821. doi: 10.1007/s00415-022-11088-w. Epub 2022 Apr 15. J Neurol. 2022. PMID: 35426533 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
