

Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure
- PMID: 34905511
- PMCID: PMC8803336
- DOI: 10.1172/JCI146926
Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure
Abstract
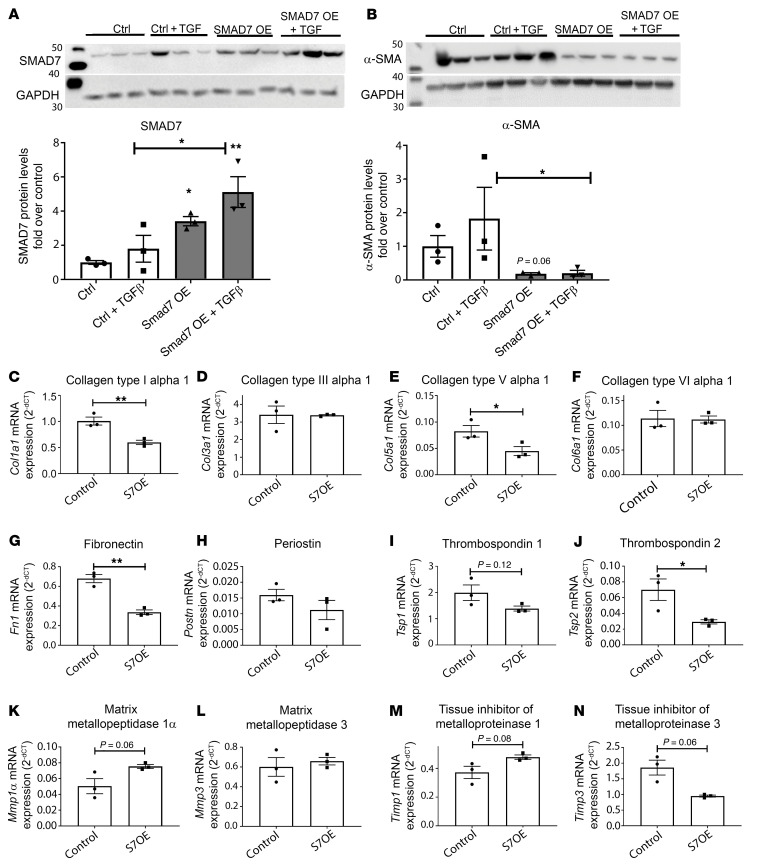
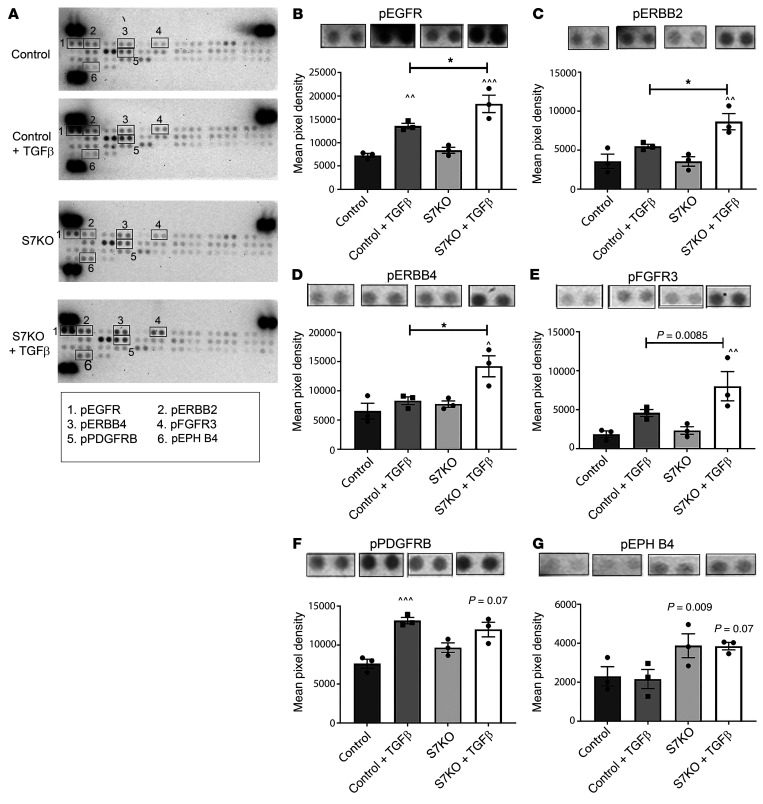
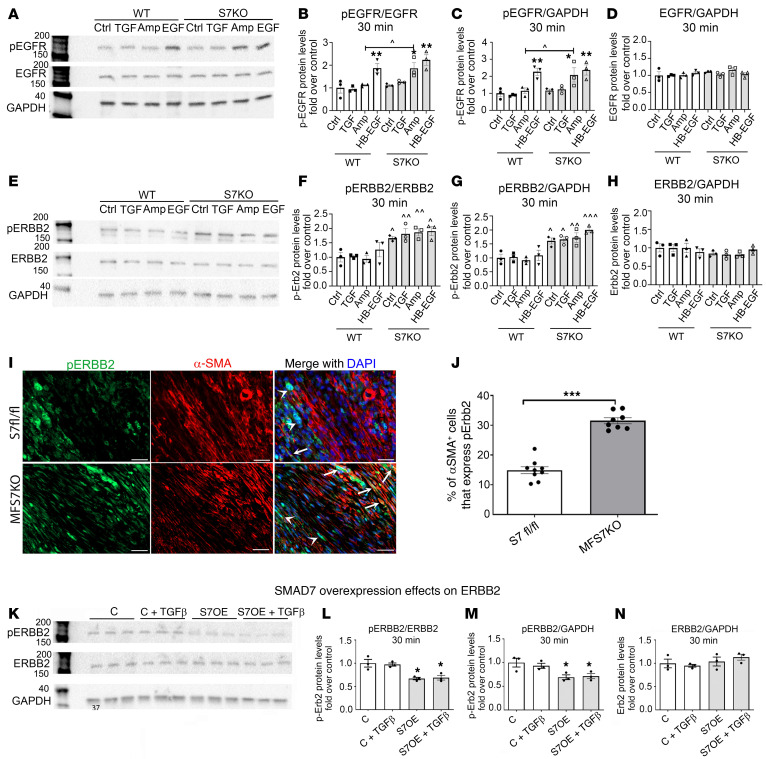
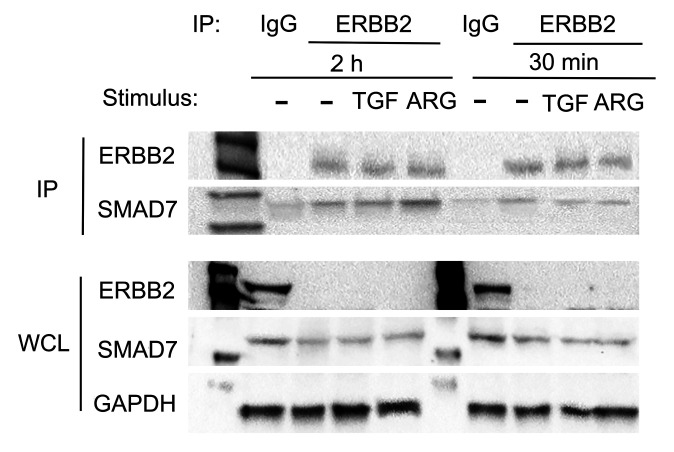
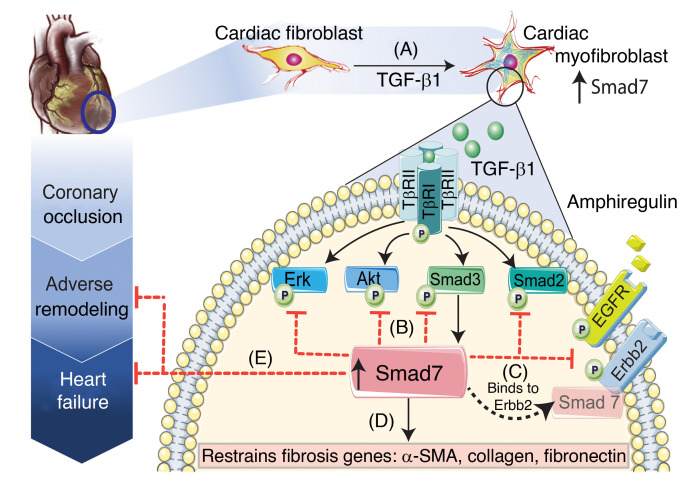
Repair of the infarcted heart requires TGF-β/Smad3 signaling in cardiac myofibroblasts. However, TGF-β-driven myofibroblast activation needs to be tightly regulated in order to prevent excessive fibrosis and adverse remodeling that may precipitate heart failure. We hypothesized that induction of the inhibitory Smad, Smad7, may restrain infarct myofibroblast activation, and we examined the molecular mechanisms of Smad7 actions. In a mouse model of nonreperfused infarction, Smad3 activation triggered Smad7 synthesis in α-SMA+ infarct myofibroblasts, but not in α-SMA-PDGFRα+ fibroblasts. Myofibroblast-specific Smad7 loss increased heart failure-related mortality, worsened dysfunction, and accentuated fibrosis in the infarct border zone and in the papillary muscles. Smad7 attenuated myofibroblast activation and reduced synthesis of structural and matricellular extracellular matrix proteins. Smad7 effects on TGF-β cascades involved deactivation of Smad2/3 and non-Smad pathways, without any effects on TGF-β receptor activity. Unbiased transcriptomic and proteomic analysis identified receptor tyrosine kinase signaling as a major target of Smad7. Smad7 interacted with ErbB2 in a TGF-β-independent manner and restrained ErbB1/ErbB2 activation, suppressing fibroblast expression of fibrogenic proteases, integrins, and CD44. Smad7 induction in myofibroblasts serves as an endogenous TGF-β-induced negative feedback mechanism that inhibits postinfarction fibrosis by restraining Smad-dependent and Smad-independent TGF-β responses, and by suppressing TGF-β-independent fibrogenic actions of ErbB2.
Keywords: Cardiology; Fibrosis; Growth factors; Heart failure; Immunology.
Conflict of interest statement
Figures
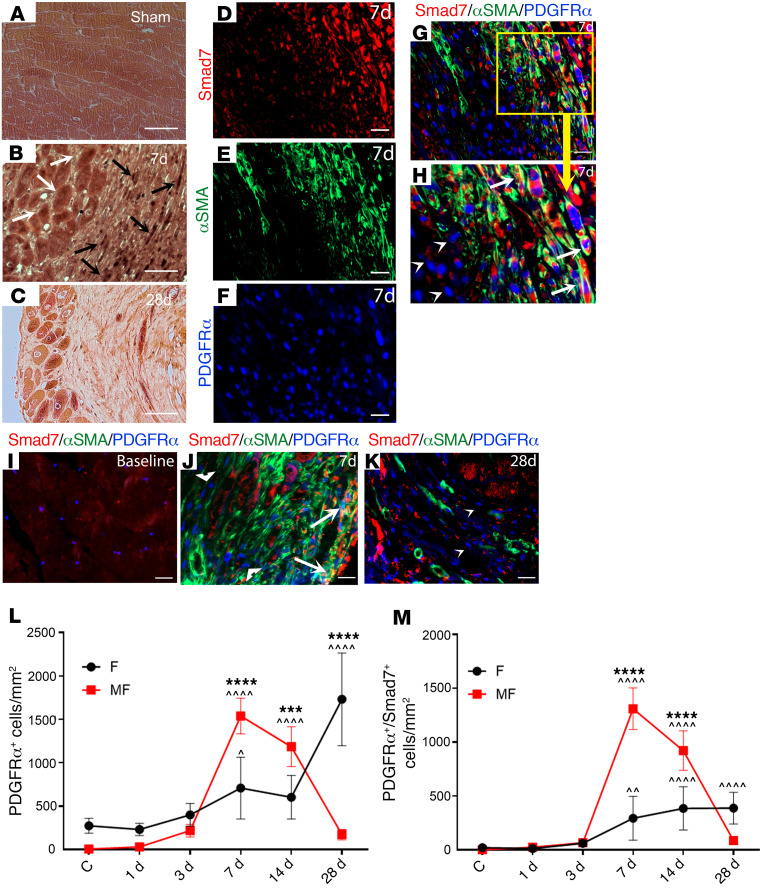
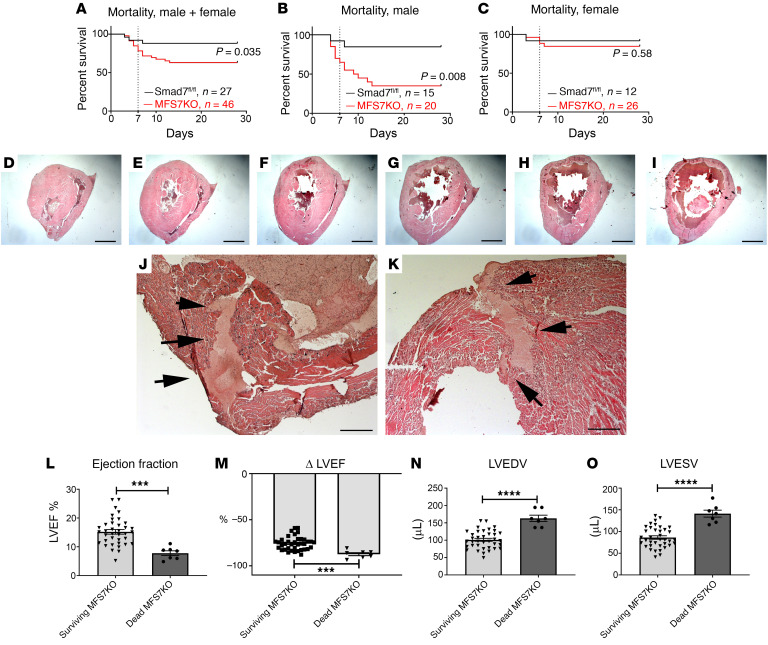

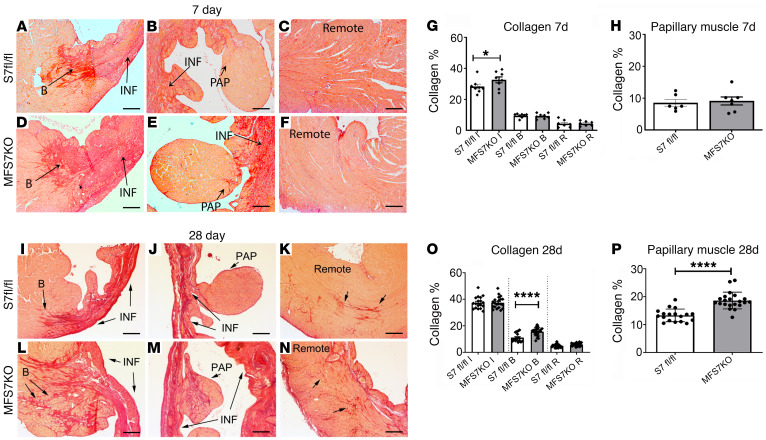
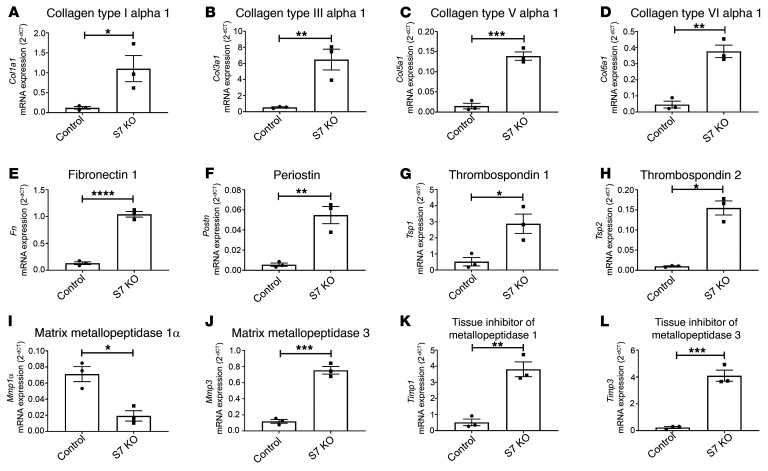


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
