

Regulatory role of cathepsin L in induction of nuclear laminopathy in Alzheimer's disease
- PMID: 34905652
- PMCID: PMC8761039
- DOI: 10.1111/acel.13531
Regulatory role of cathepsin L in induction of nuclear laminopathy in Alzheimer's disease
Abstract
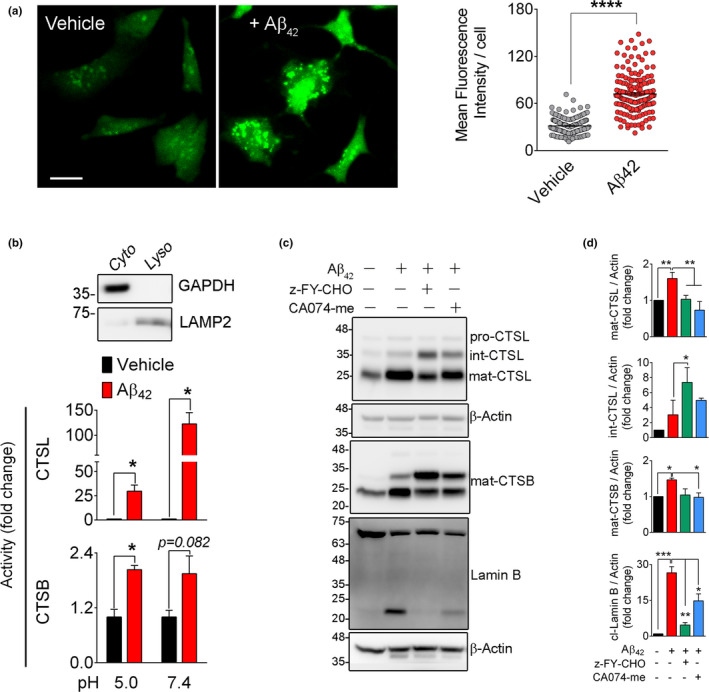
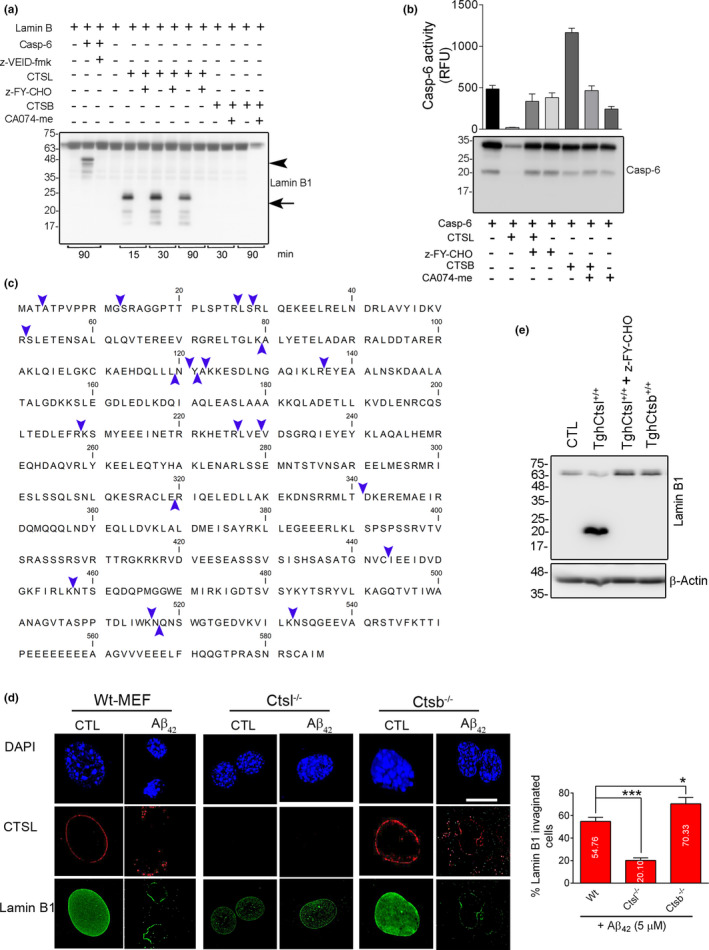
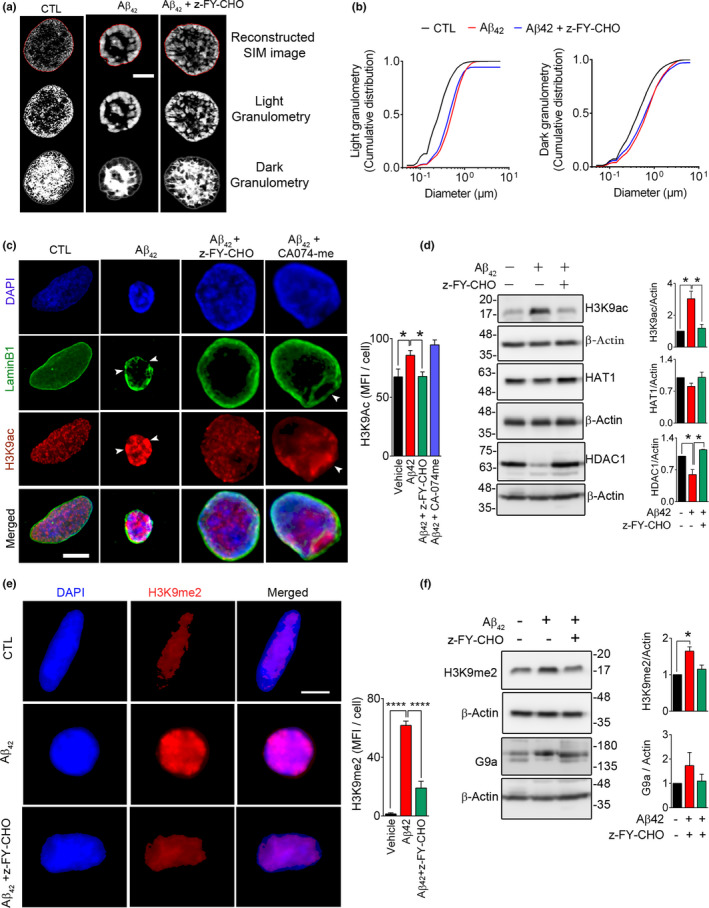
Experimental and clinical therapies in the field of Alzheimer's disease (AD) have focused on elimination of extracellular amyloid beta aggregates or prevention of cytoplasmic neuronal fibrillary tangles formation, yet these approaches have been generally ineffective. Interruption of nuclear lamina integrity, or laminopathy, is a newly identified concept in AD pathophysiology. Unraveling the molecular players in the induction of nuclear lamina damage may lead to identification of new therapies. Here, using 3xTg and APP/PS1 mouse models of AD, and in vitro model of amyloid beta42 (Aβ42) toxicity in primary neuronal cultures and SH-SY5Y neuroblastoma cells, we have uncovered a key role for cathepsin L in the induction of nuclear lamina damage. The applicability of our findings to AD pathophysiology was validated in brain autopsy samples from patients. We report that upregulation of cathepsin L is an important process in the induction of nuclear lamina damage, shown by lamin B1 cleavage, and is associated with epigenetic modifications in AD pathophysiology. More importantly, pharmacological targeting and genetic knock out of cathepsin L mitigated Aβ42 induced lamin B1 degradation and downstream structural and molecular changes. Affirming these findings, overexpression of cathepsin L alone was sufficient to induce lamin B1 cleavage. The proteolytic activity of cathepsin L on lamin B1 was confirmed using mass spectrometry. Our research identifies cathepsin L as a newly identified lamin B1 protease and mediator of laminopathy observed in AD. These results uncover a new aspect in the pathophysiology of AD that can be pharmacologically prevented, raising hope for potential therapeutic interventions.
Keywords: acetylation; amyloid beta; chromatin; histone; lysosomal membrane permeabilization; methylation; nuclear lamina; super-resolution microscopy.
© 2021 The Authors. Aging Cell published by Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
All the authors declare no competing of interests.
Figures
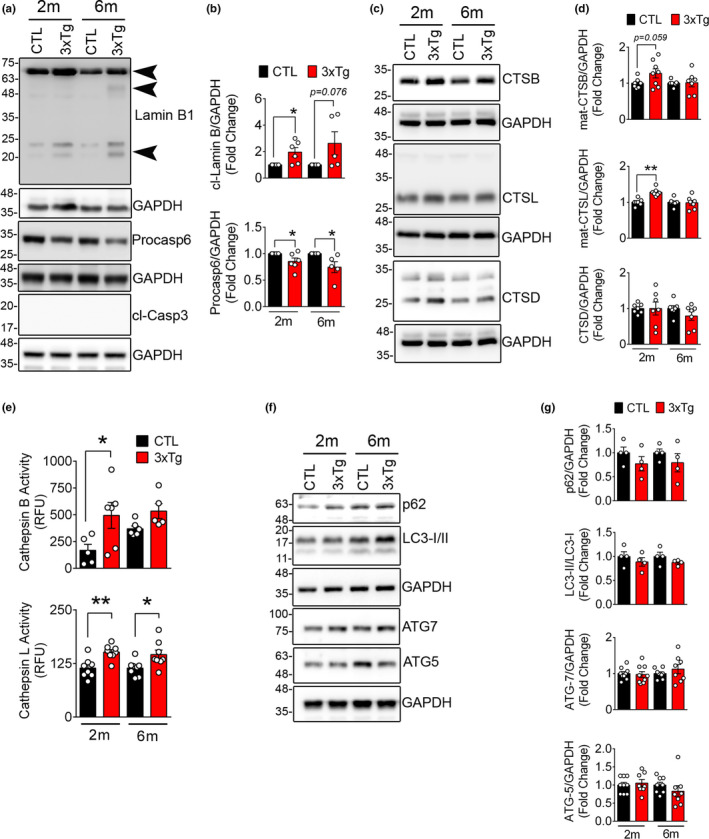
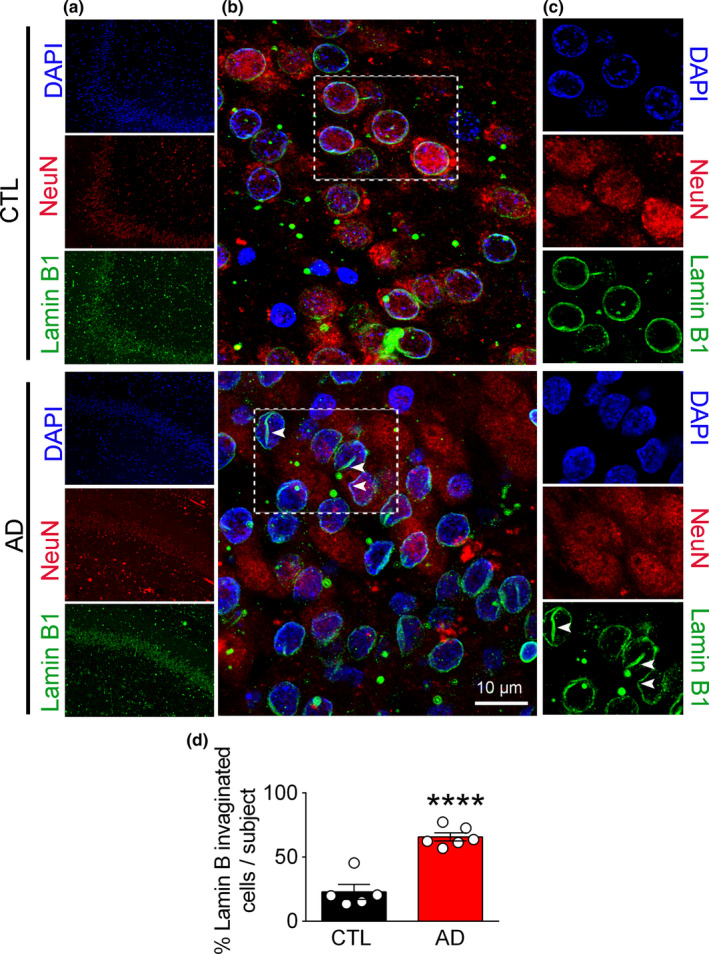
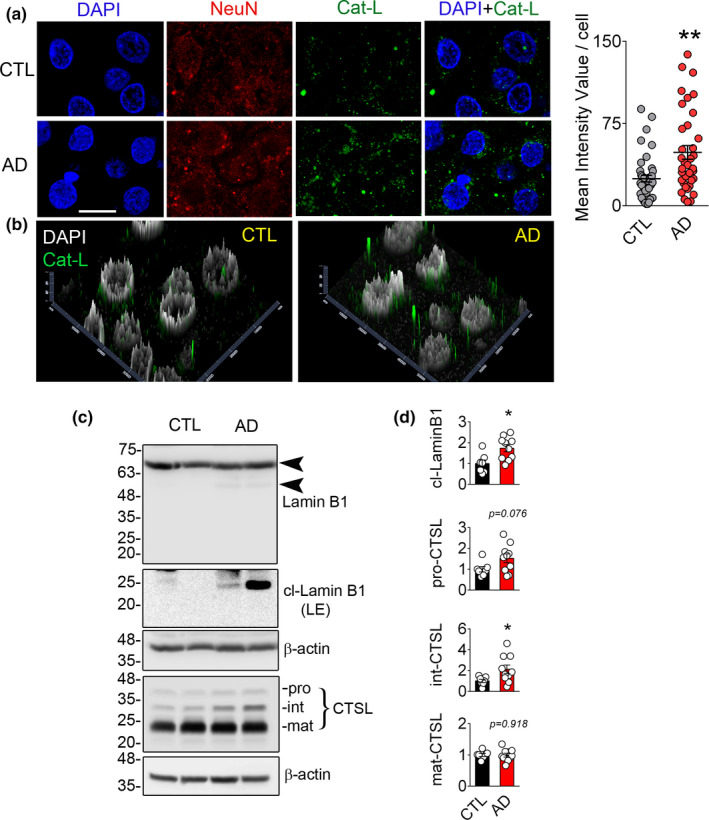
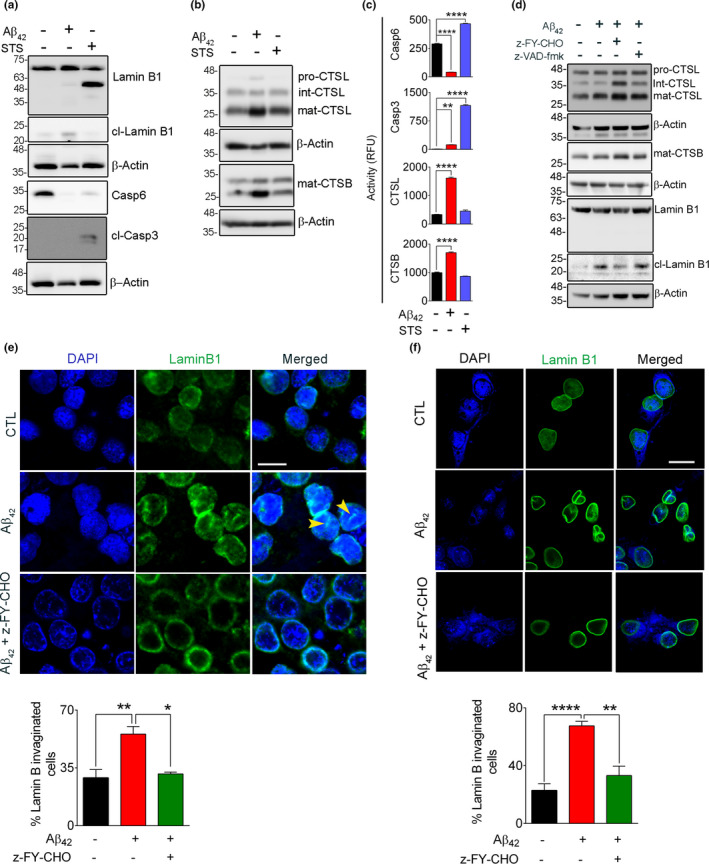
References
-
- Bertero, A. , Fields, P. A. , Smith, A. S. T. , Leonard, A. , Beussman, K. , Sniadecki, N. J. , Kim, D.‐H. , Tse, H.‐F. , Pabon, L. , Shendure, J. , Noble, W. S. , & Murry, C. E. (2019). Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. Journal of Cell Biology, 218(9), 2919–2944. 10.1083/jcb.201902117 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
