

Benchmarking UMI-based single-cell RNA-seq preprocessing workflows
- PMID: 34906205
- PMCID: PMC8672463
- DOI: 10.1186/s13059-021-02552-3
Benchmarking UMI-based single-cell RNA-seq preprocessing workflows
Abstract
Background: Single-cell RNA-sequencing (scRNA-seq) technologies and associated analysis methods have rapidly developed in recent years. This includes preprocessing methods, which assign sequencing reads to genes to create count matrices for downstream analysis. While several packaged preprocessing workflows have been developed to provide users with convenient tools for handling this process, how they compare to one another and how they influence downstream analysis have not been well studied.
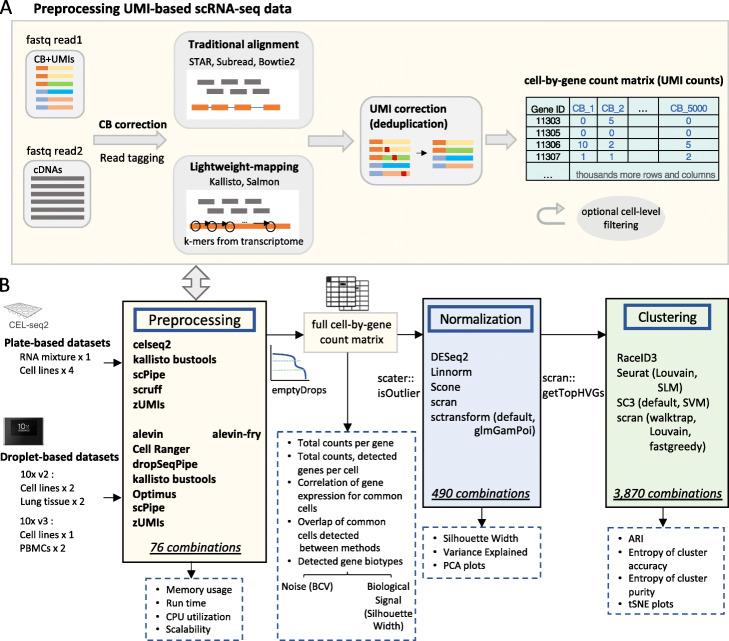
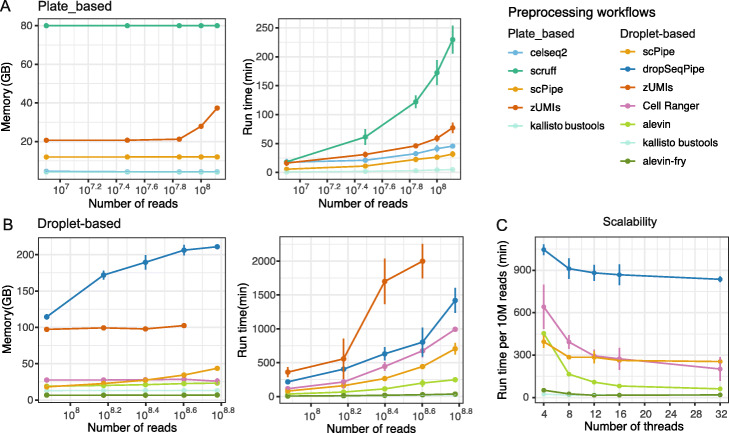
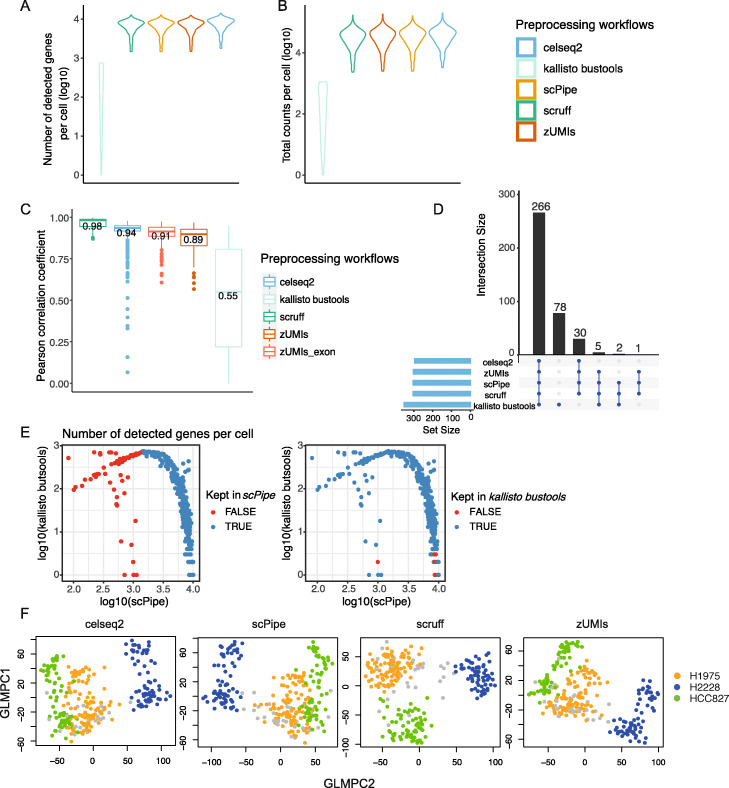
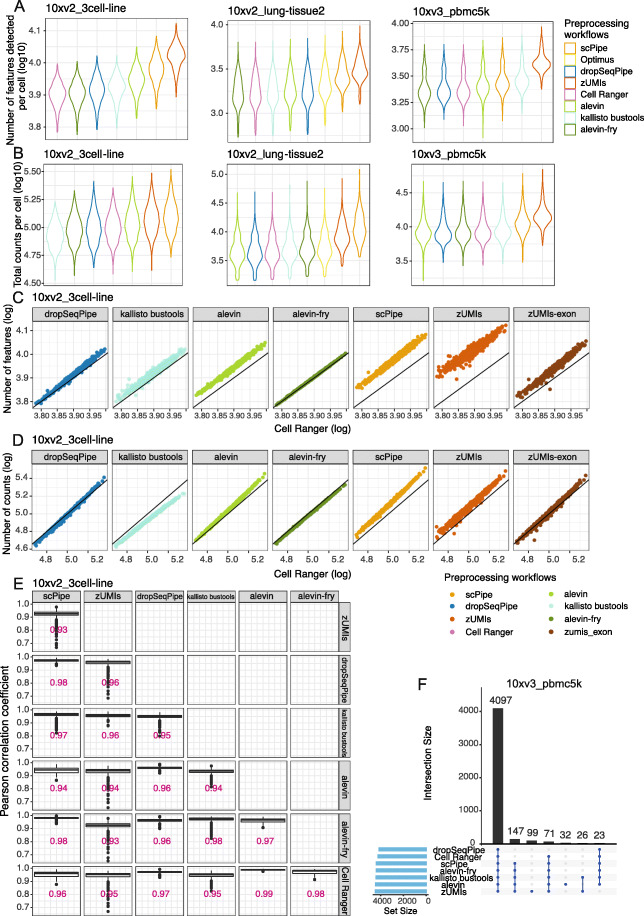
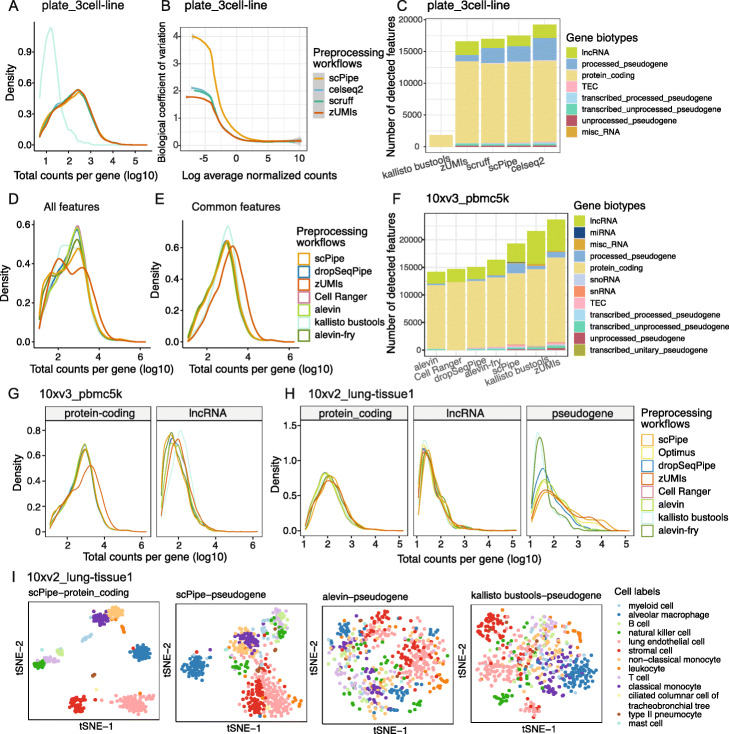
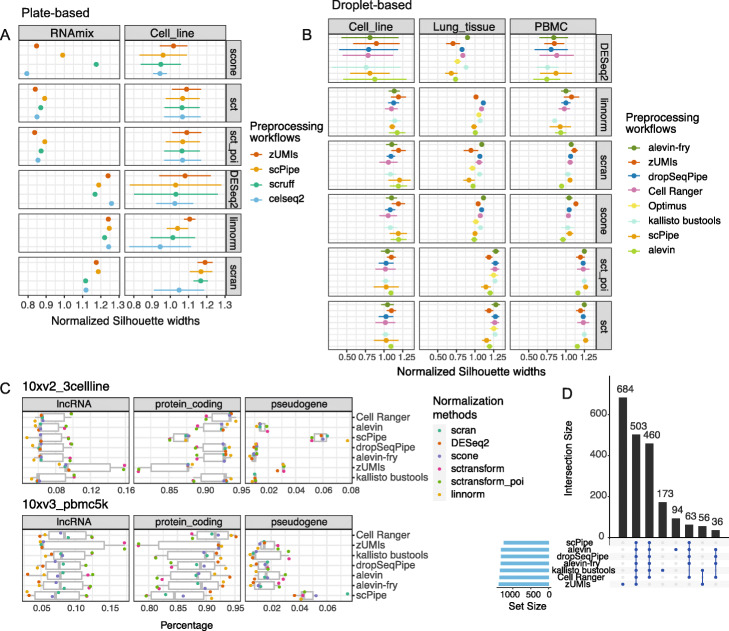
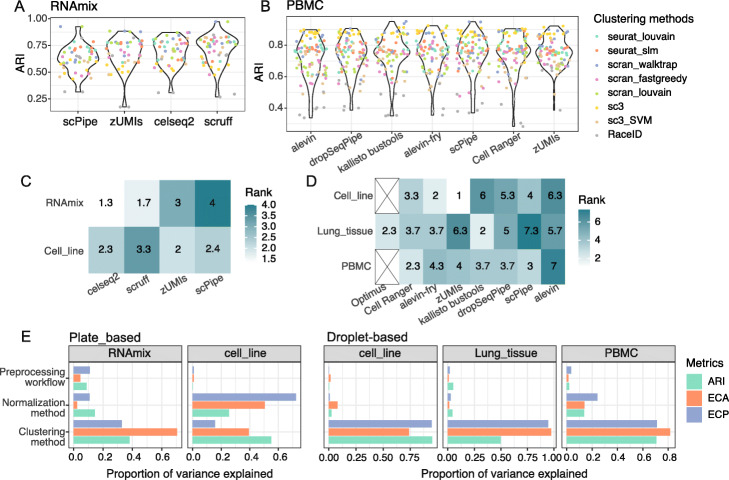
Results: Here, we systematically benchmark the performance of 10 end-to-end preprocessing workflows (Cell Ranger, Optimus, salmon alevin, alevin-fry, kallisto bustools, dropSeqPipe, scPipe, zUMIs, celseq2, and scruff) using datasets yielding different biological complexity levels generated by CEL-Seq2 and 10x Chromium platforms. We compare these workflows in terms of their quantification properties directly and their impact on normalization and clustering by evaluating the performance of different method combinations. While the scRNA-seq preprocessing workflows compared vary in their detection and quantification of genes across datasets, after downstream analysis with performant normalization and clustering methods, almost all combinations produce clustering results that agree well with the known cell type labels that provided the ground truth in our analysis.
Conclusions: In summary, the choice of preprocessing method was found to be less important than other steps in the scRNA-seq analysis process. Our study comprehensively compares common scRNA-seq preprocessing workflows and summarizes their characteristics to guide workflow users.
Keywords: Methods comparison; Preprocessing; Sequencing analysis; Transcriptomics; scRNA-seq.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Svensson V, Vento-Tormo R, Teichmann S. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13(4):599–604. - PubMed
-
- Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. 2019;37(5):547–54. - PubMed
-
- Tian L, Dong X, Freytag S, Lê Cao K, Su S, JalalAbadi A, Amann-Zalcenstein D, Weber T, Seidi A, Jabbari J, Naik S, Ritchie M. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods. 2019;16(6):479–87. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
