

The importance of automation in genetic diagnosis: Lessons from analyzing an inherited retinal degeneration cohort with the Mendelian Analysis Toolkit (MATK)
- PMID: 34906470
- PMCID: PMC9200473
- DOI: 10.1016/j.gim.2021.09.015
The importance of automation in genetic diagnosis: Lessons from analyzing an inherited retinal degeneration cohort with the Mendelian Analysis Toolkit (MATK)
Abstract
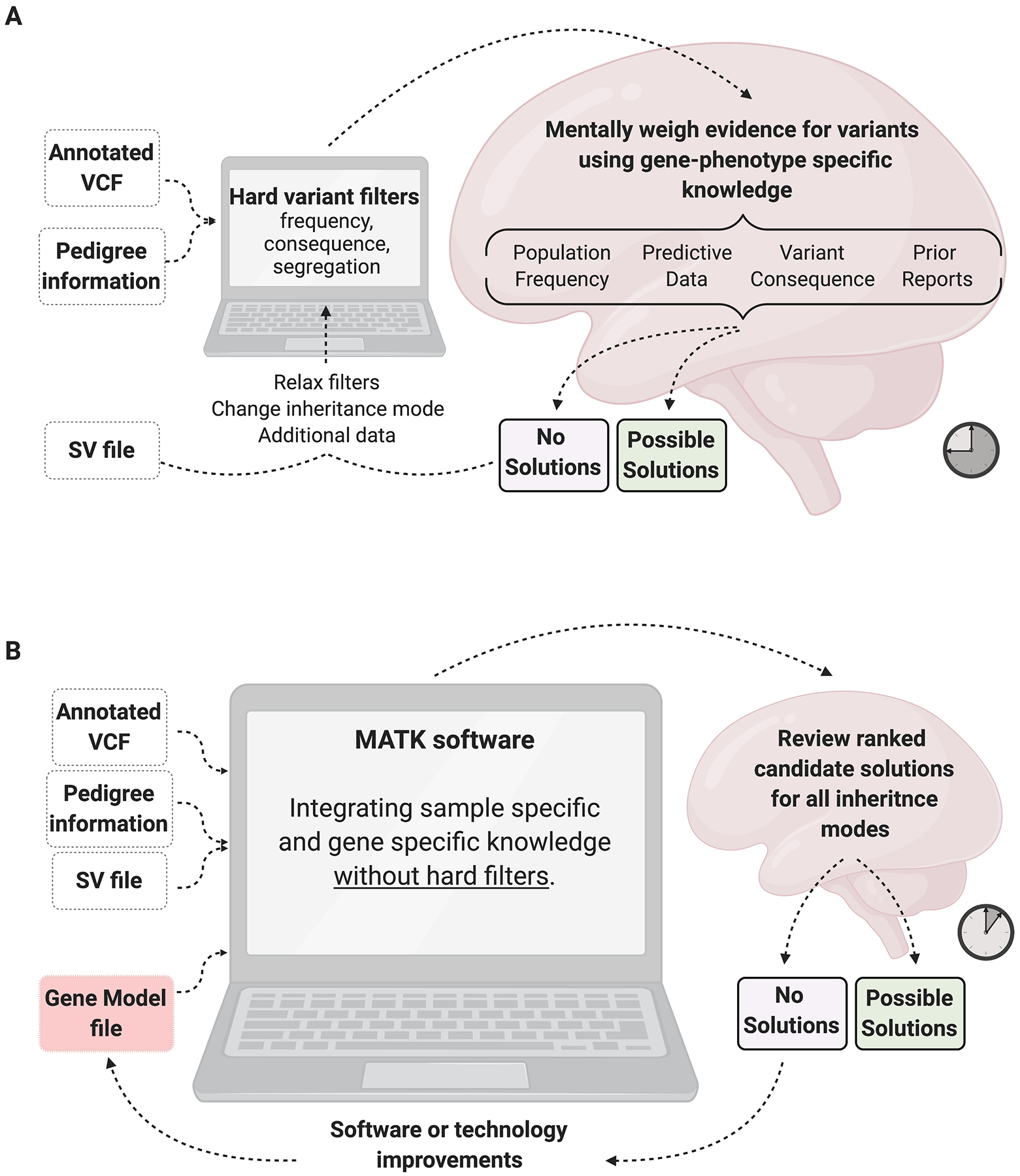
Purpose: In Mendelian disease diagnosis, variant analysis is a repetitive, error-prone, and time consuming process. To address this, we have developed the Mendelian Analysis Toolkit (MATK), a configurable, automated variant ranking program.
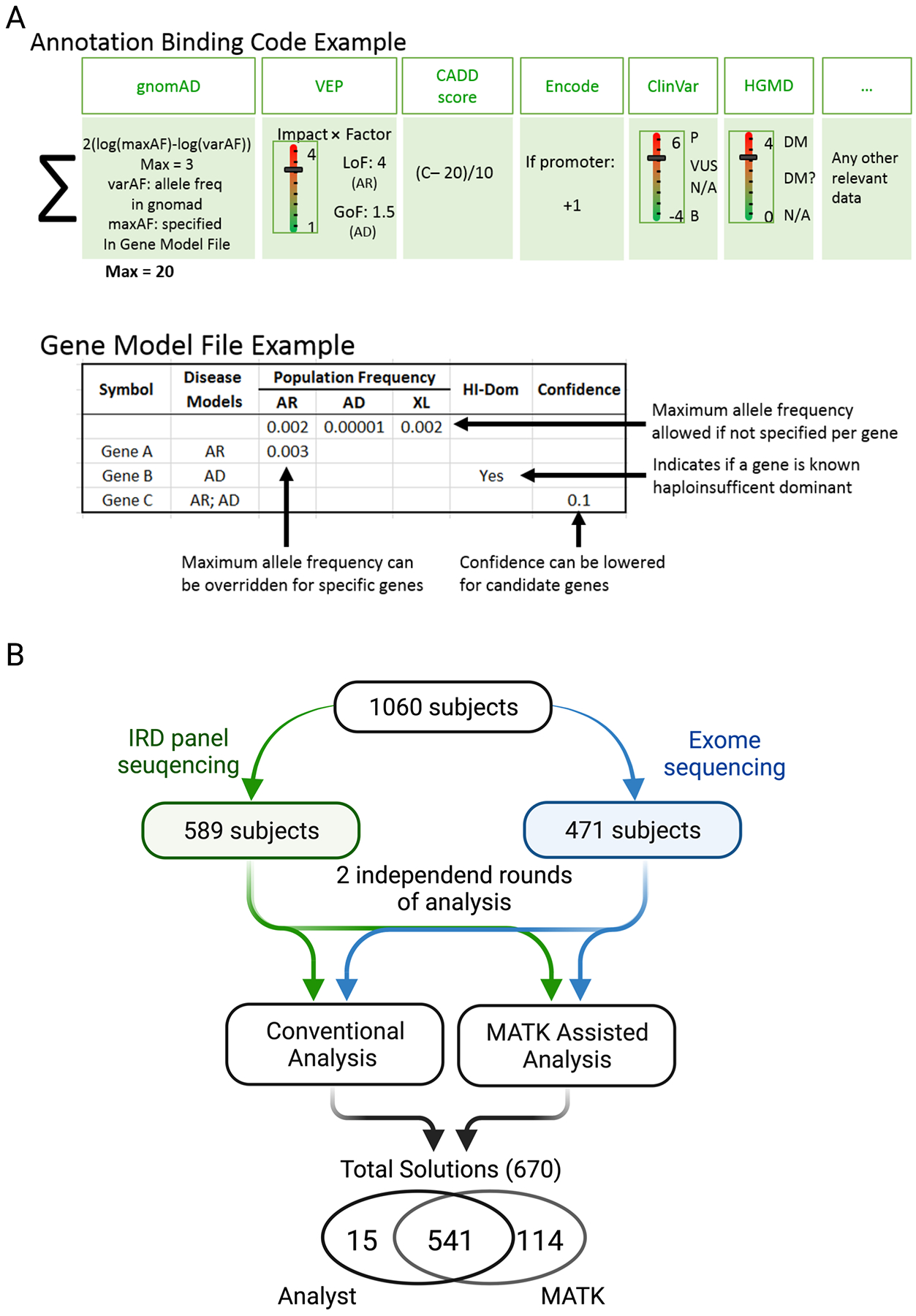
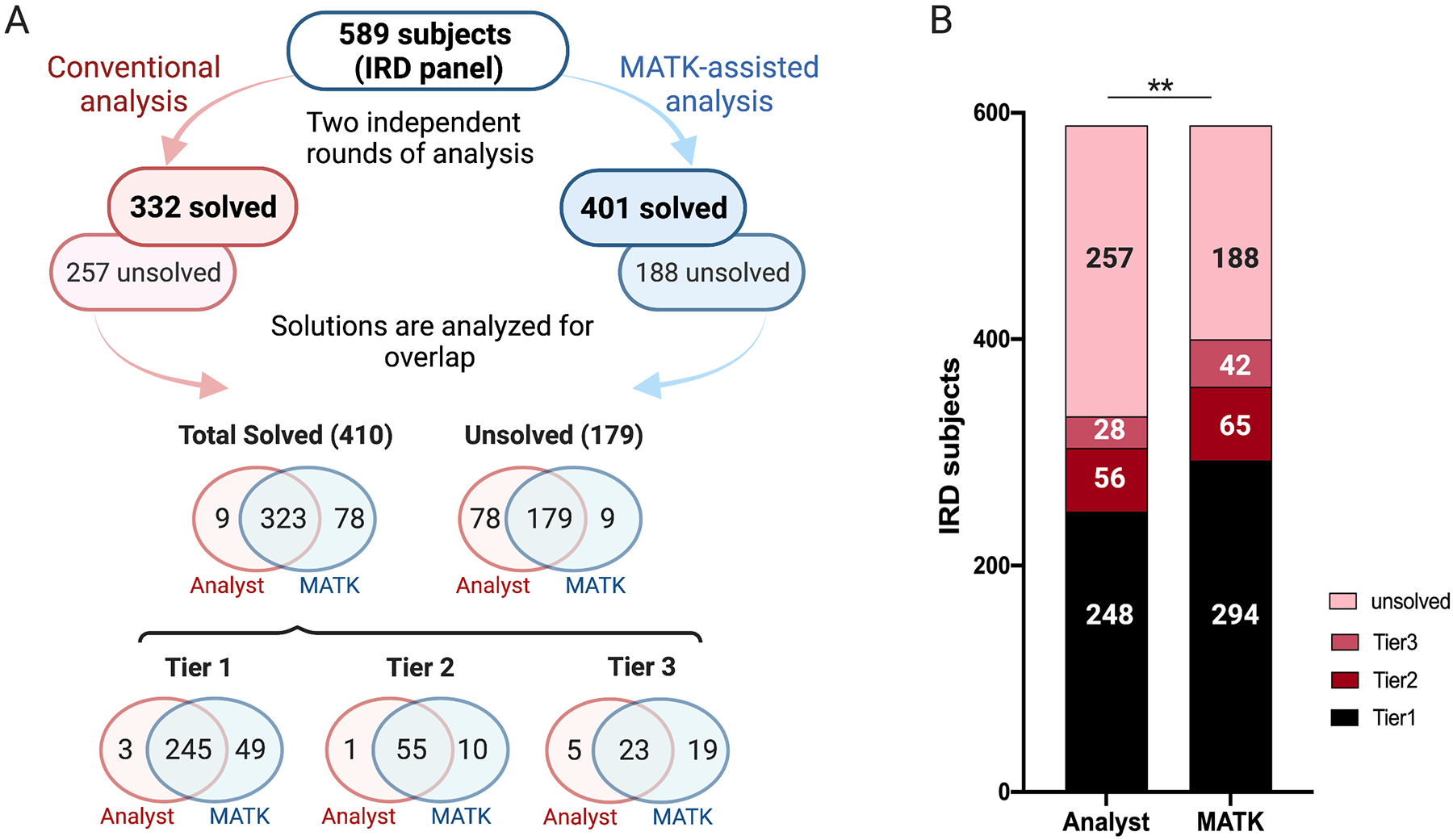
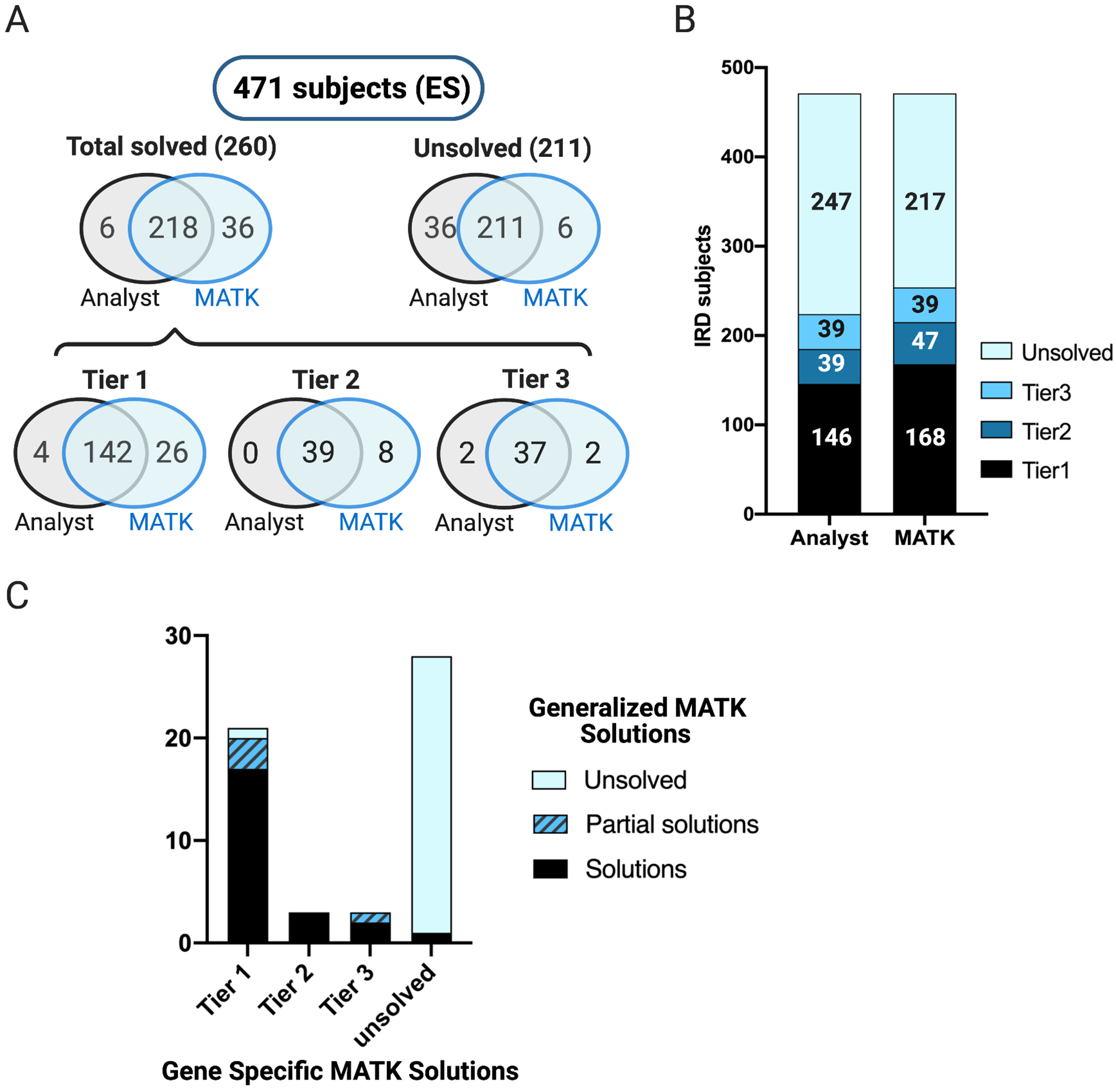
Methods: MATK aggregates variant information from multiple annotation sources and uses expert-designed rules with parameterized weights to produce a ranked list of potentially causal solutions. MATK performance was measured by a comparison between MATK-aided and human-domain expert analyses of 1060 families with inherited retinal degeneration (IRD), analyzed using an IRD-specific gene panel (589 individuals) and exome sequencing (471 families).
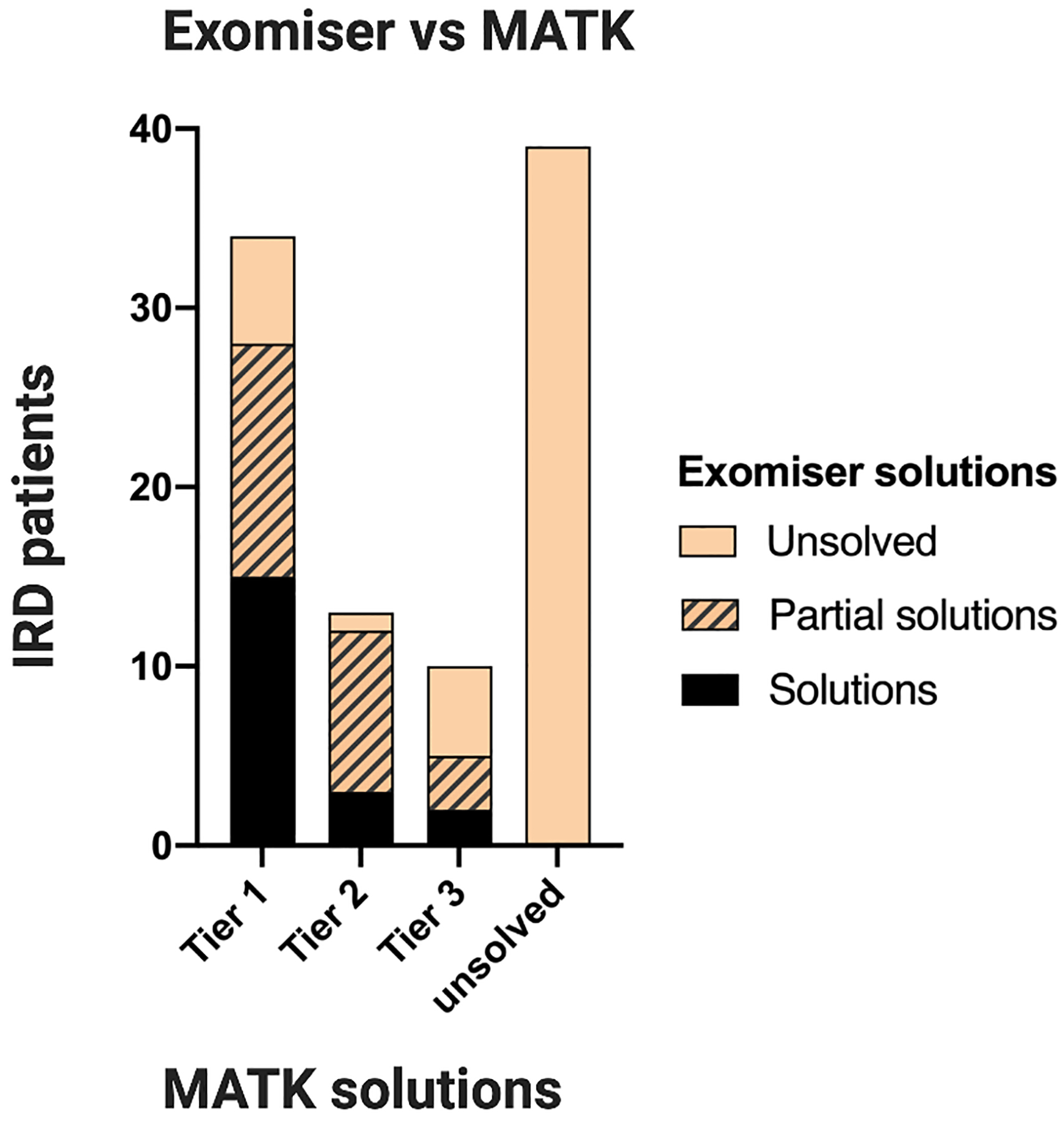
Results: When comparing MATK-assisted analysis with expert curation in both the IRD-specific gene panel and exome sequencing (1060 subjects), 97.3% of potential solutions found by experts were also identified by the MATK-assisted analysis (541 solutions identified with MATK of 556 solutions found by conventional analysis). Furthermore, MATK-assisted analysis identified 114 additional potential solutions from the 504 cases unsolved by conventional analysis.
Conclusion: MATK expedites the process of identification of likely solving variants in Mendelian traits, and reduces variability stemming from human error and researcher bias. MATK facilitates data reanalysis to keep up with the constantly improving annotation sources and next-generation sequencing processing pipelines. The software is open source and available at https://gitlab.com/matthew_maher/mendelanalysis.
Keywords: Automation; Mendelian analysis; Variant ranking.
Copyright © 2021 American College of Medical Genetics and Genomics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest The authors declare no conflict of interest related to the work presented in this manuscript. Naomi E. Wagner is currently a full-time employee of and owns stocks in Invitae Corporation; however, none of the work she did on this study was done while at the company.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
