

Rickets guidance: part I-diagnostic workup
- PMID: 34910242
- PMCID: PMC9307538
- DOI: 10.1007/s00467-021-05328-w
Rickets guidance: part I-diagnostic workup
Abstract
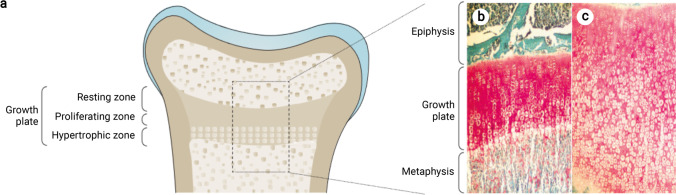
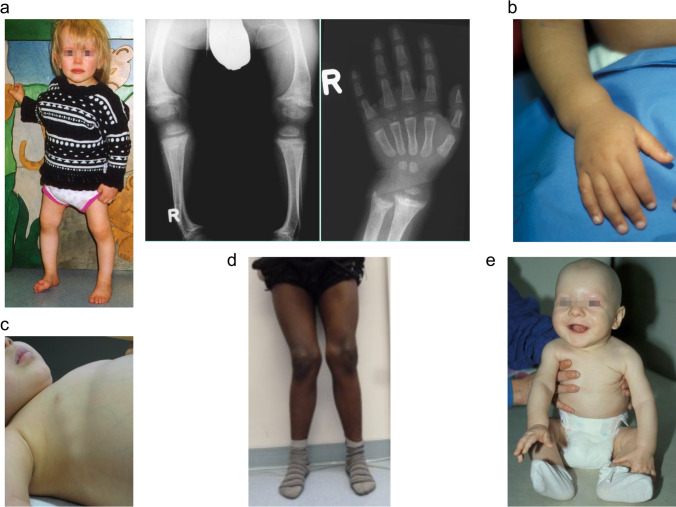
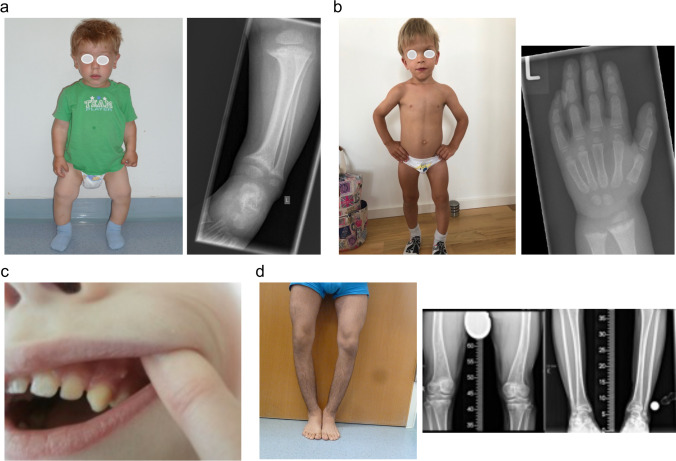
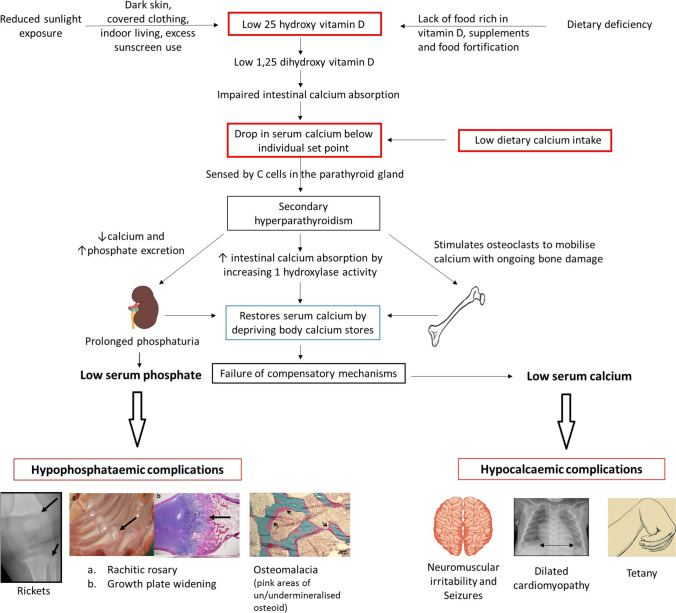
Rickets is a disease of the growing child arising from alterations in calcium and phosphate homeostasis resulting in impaired apoptosis of hypertrophic chondrocytes in the growth plate. Its symptoms depend on the patients' age, duration of disease, and underlying disorder. Common features include thickened wrists and ankles due to widened metaphyses, growth failure, bone pain, muscle weakness, waddling gait, and leg bowing. Affected infants often show delayed closure of the fontanelles, frontal bossing, and craniotabes. The diagnosis of rickets is based on the presence of these typical clinical symptoms and radiological findings on X-rays of the wrist or knee, showing metaphyseal fraying and widening of growth plates, in conjunction with elevated serum levels of alkaline phosphatase. Nutritional rickets due to vitamin D deficiency and/or dietary calcium deficiency is the most common cause of rickets. Currently, more than 20 acquired or hereditary causes of rickets are known. The latter are due to mutations in genes involved in vitamin D metabolism or action, renal phosphate reabsorption, or synthesis, or degradation of the phosphaturic hormone fibroblast growth factor 23 (FGF23). There is a substantial overlap in the clinical features between the various entities, requiring a thorough workup using biochemical analyses and, if necessary, genetic tests. Part I of this review focuses on the etiology, pathophysiology and clinical findings of rickets followed by the presentation of a diagnostic approach for correct diagnosis. Part II focuses on the management of rickets, including new therapeutic approaches based on recent clinical practice guidelines.
Keywords: Fibroblast growth factor 23; Nutritional rickets; Osteomalacia; Rickets; Vitamin D; Vitamin D-dependent rickets; X-linked hypophosphatemia.
© 2021. The Author(s).
Conflict of interest statement
D.H. and D.S. received speaker fees, consultation fees, and research grants from Kyowa Kirin. All other authors declare no conflict of interest.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
