

Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition)
- PMID: 34910301
- PMCID: PMC11115438
- DOI: 10.1002/eji.202170126
Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition)
Abstract
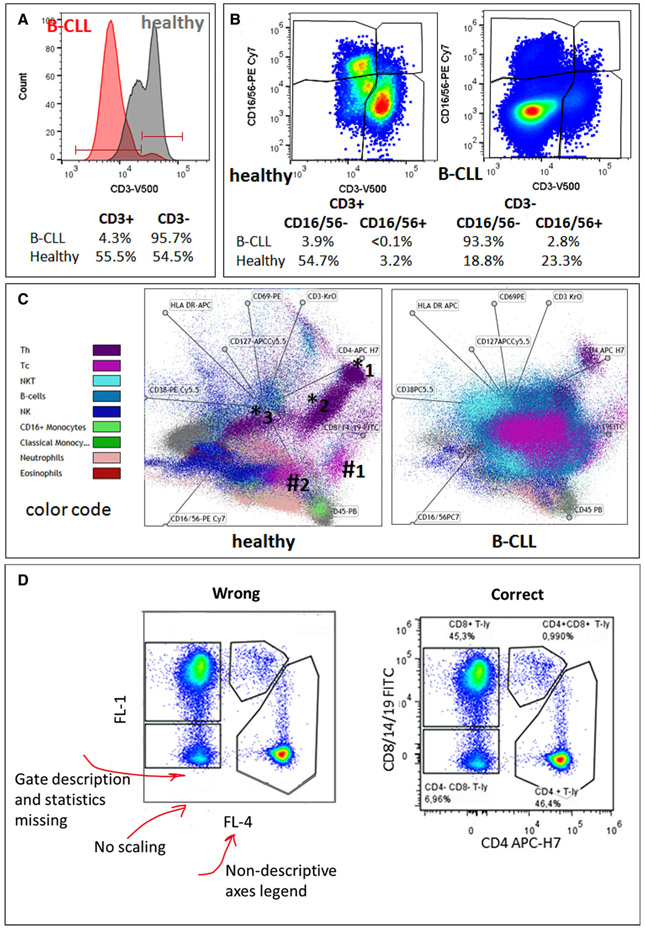
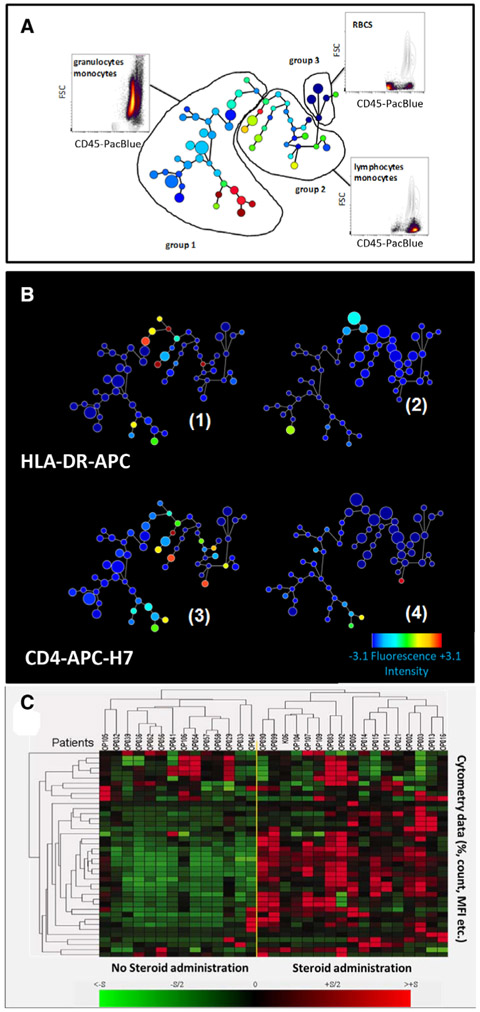
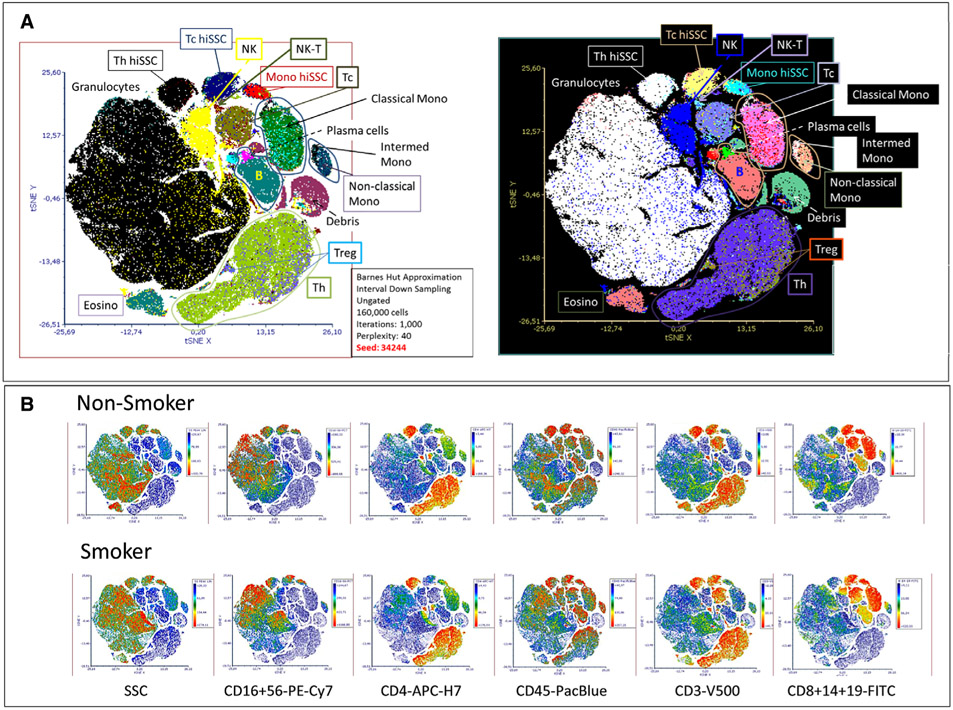
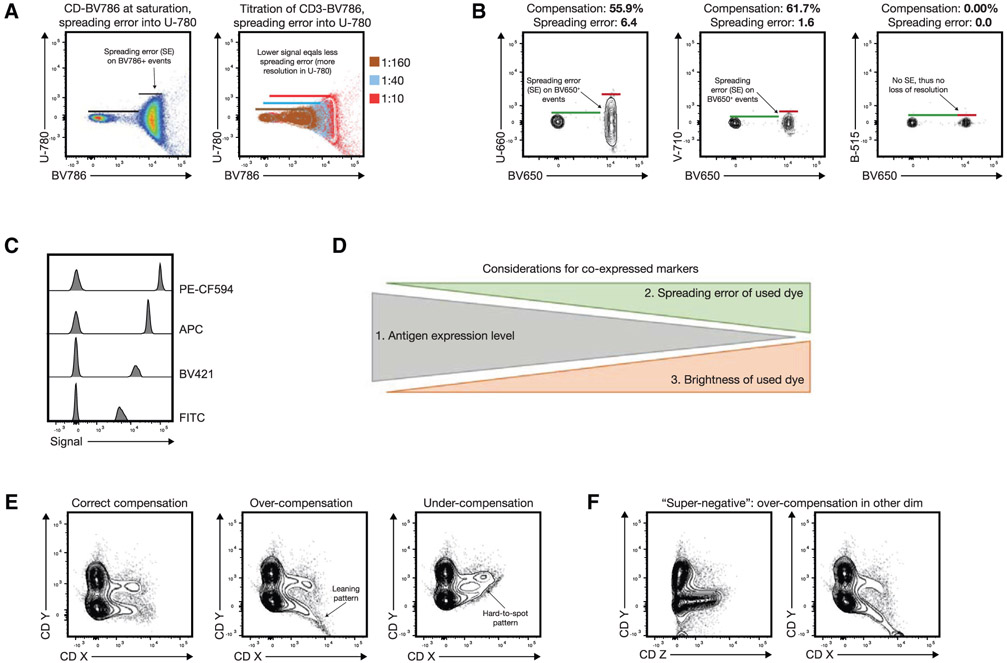
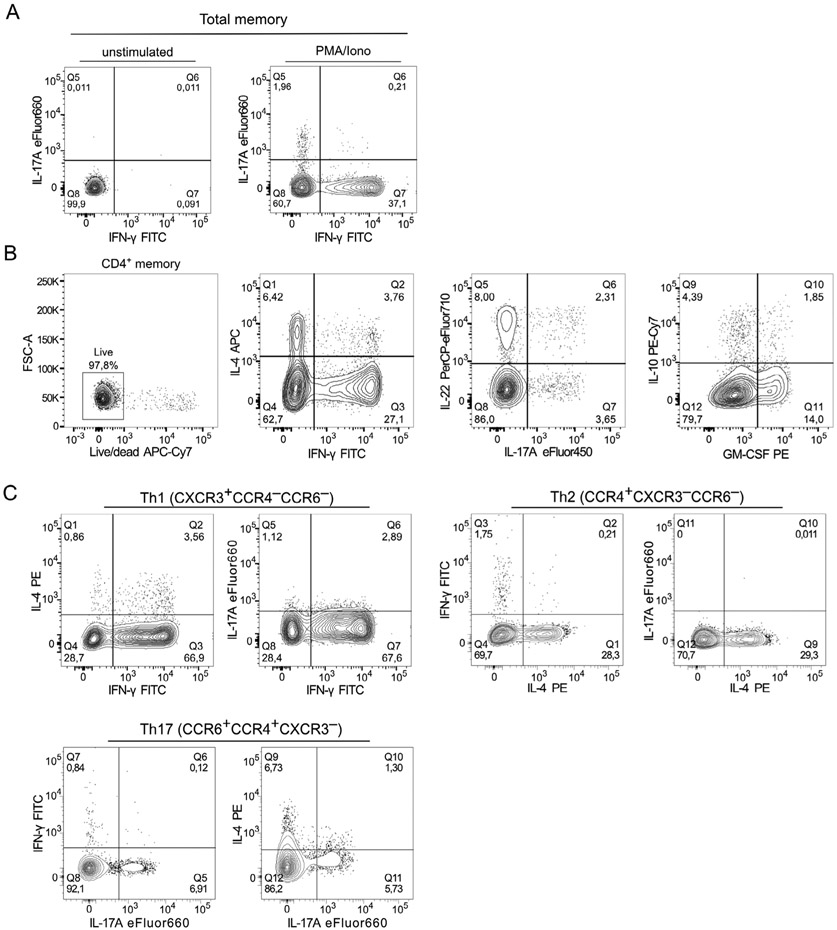
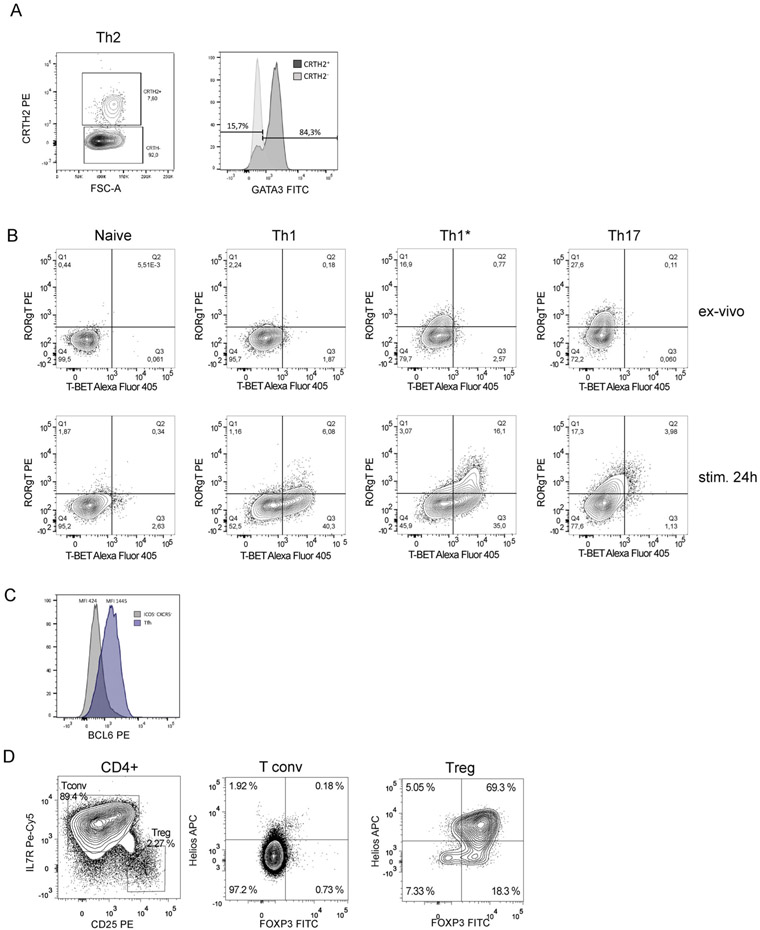
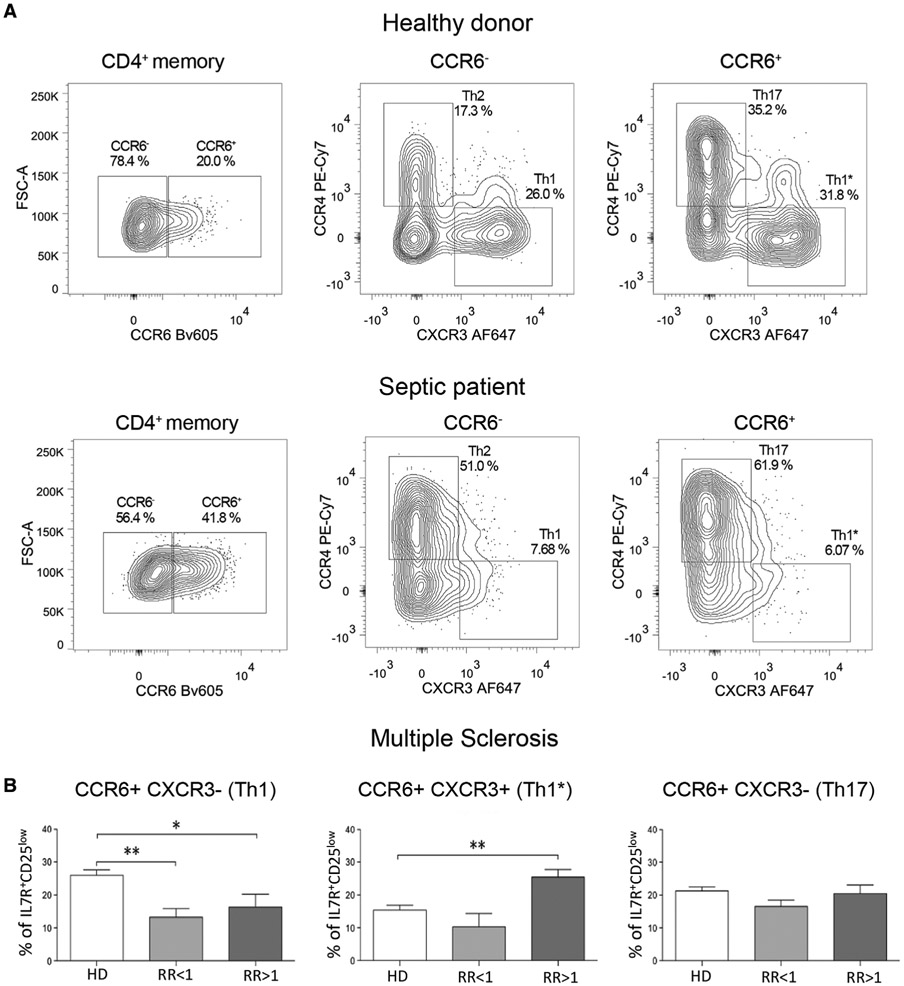
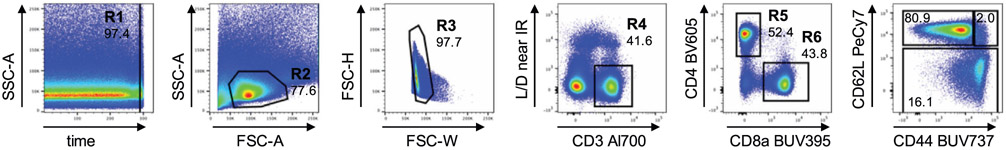
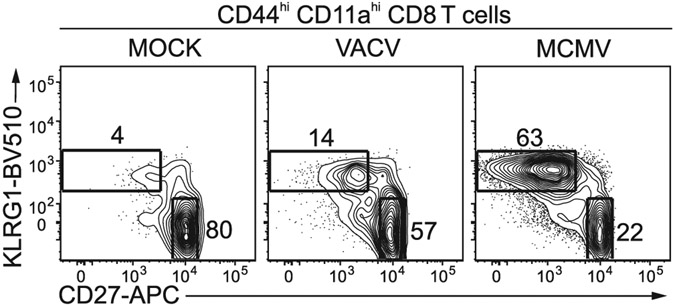
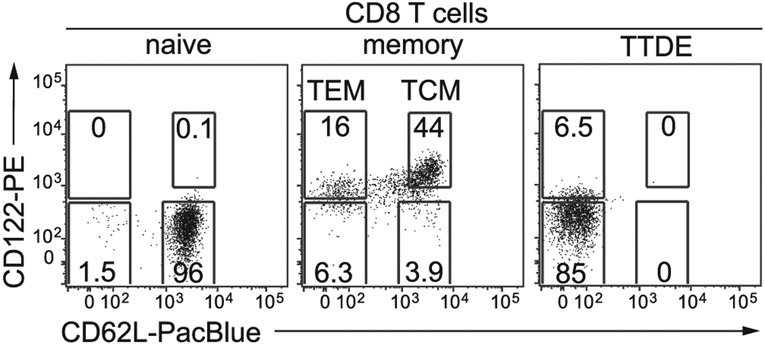
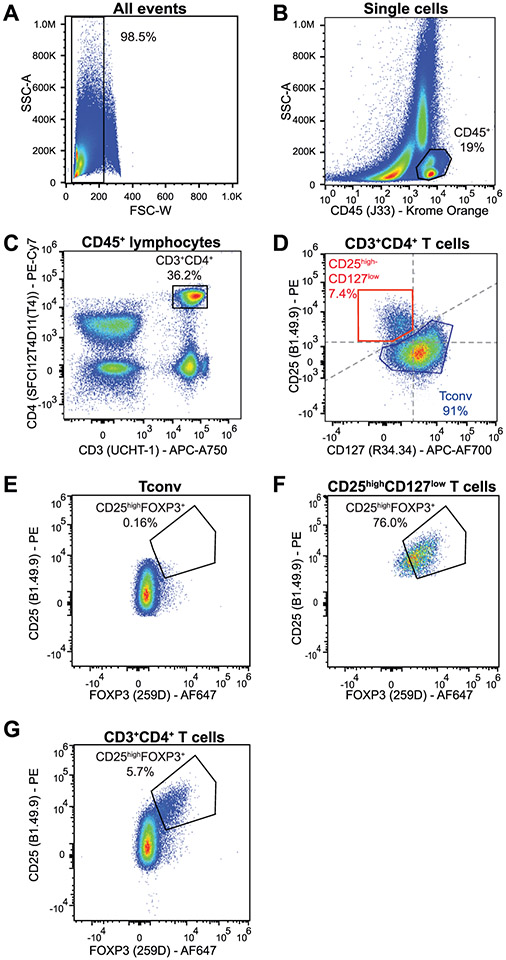
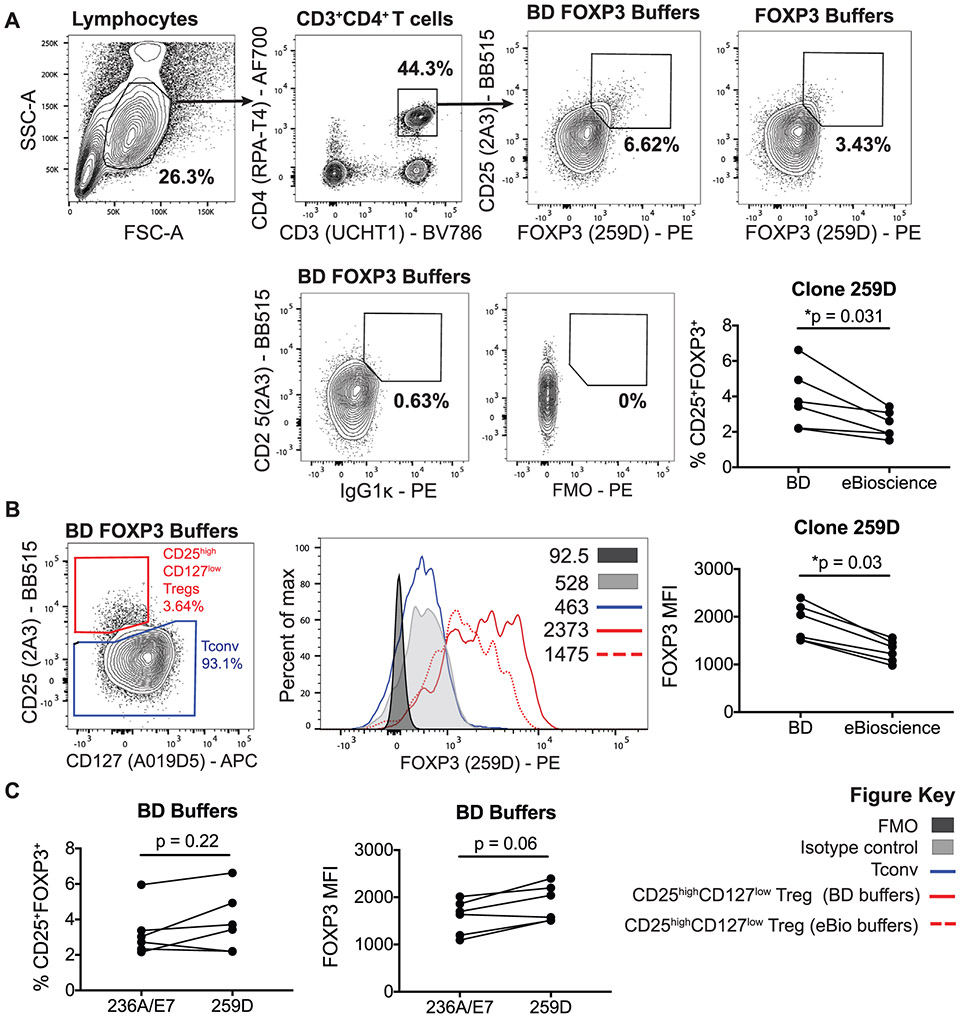
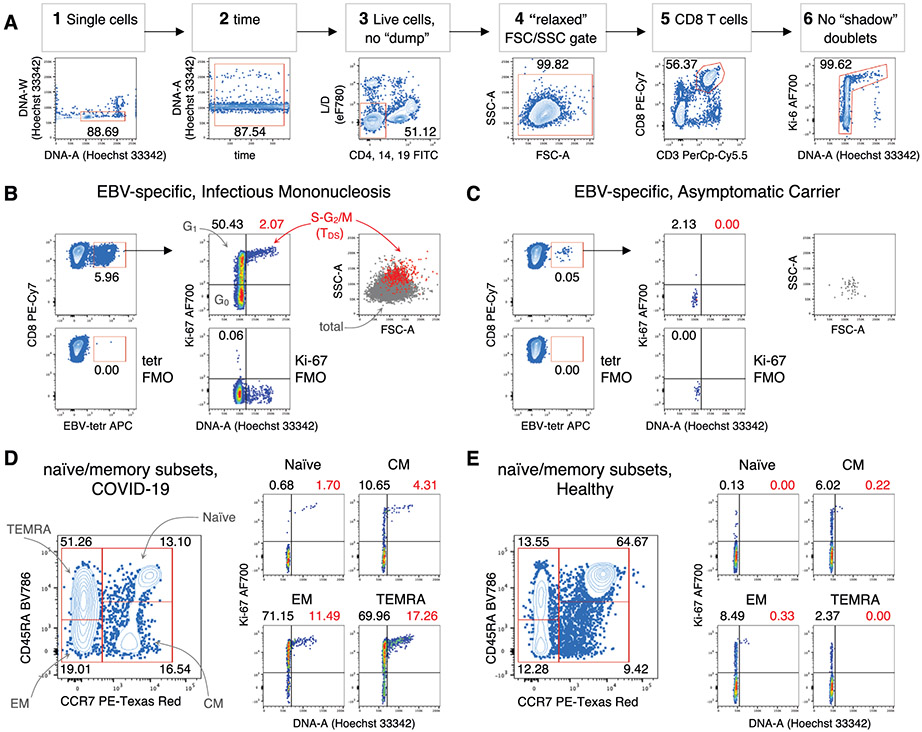
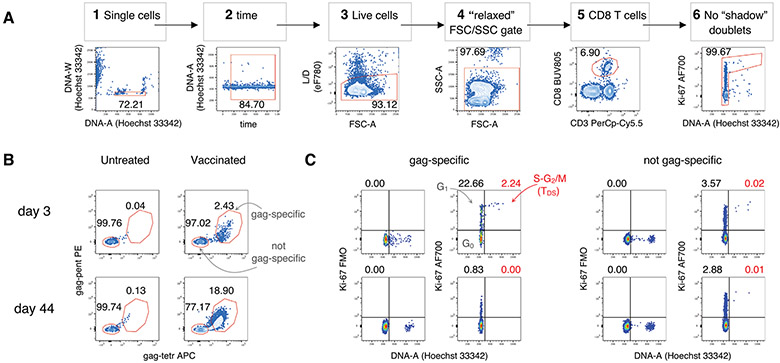
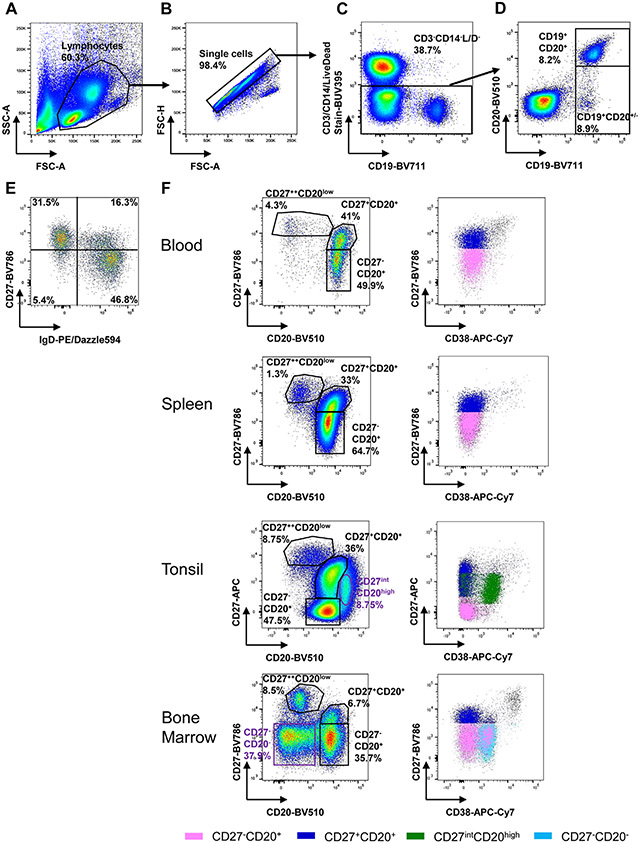
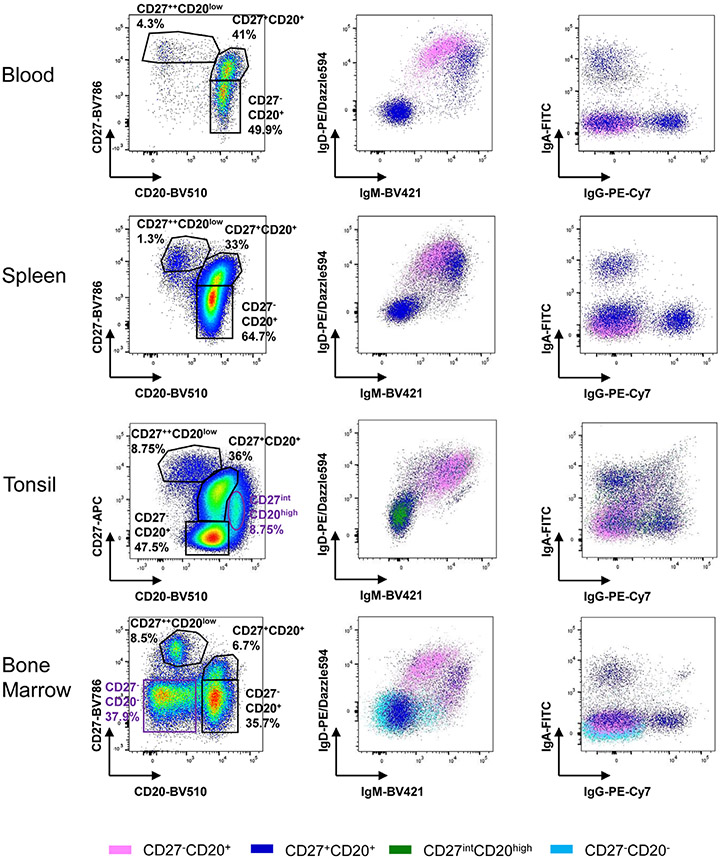
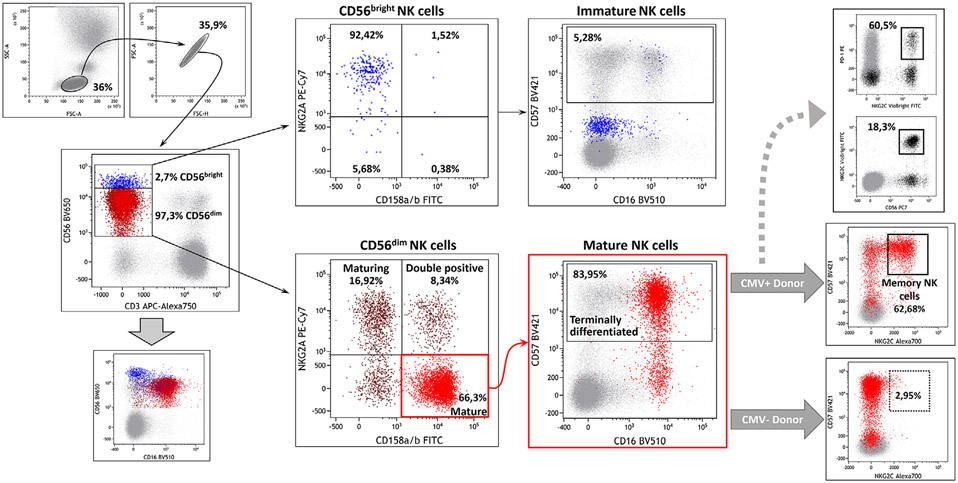
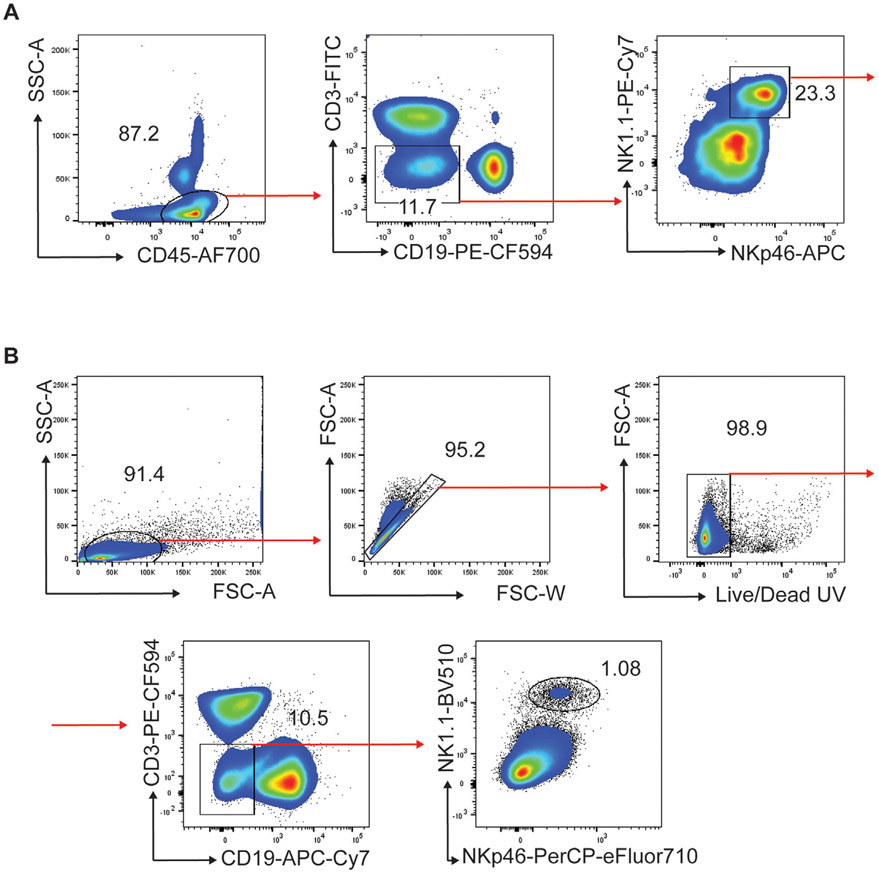
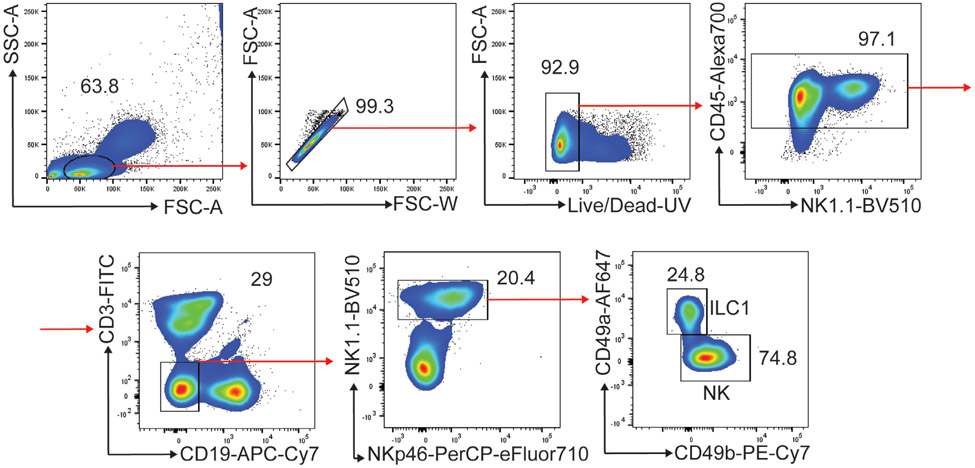
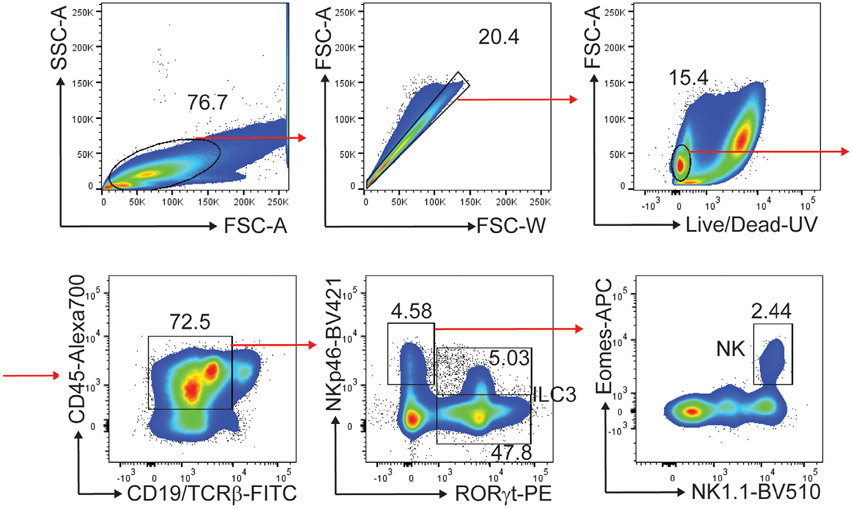
The third edition of Flow Cytometry Guidelines provides the key aspects to consider when performing flow cytometry experiments and includes comprehensive sections describing phenotypes and functional assays of all major human and murine immune cell subsets. Notably, the Guidelines contain helpful tables highlighting phenotypes and key differences between human and murine cells. Another useful feature of this edition is the flow cytometry analysis of clinical samples with examples of flow cytometry applications in the context of autoimmune diseases, cancers as well as acute and chronic infectious diseases. Furthermore, there are sections detailing tips, tricks and pitfalls to avoid. All sections are written and peer-reviewed by leading flow cytometry experts and immunologists, making this edition an essential and state-of-the-art handbook for basic and clinical researchers.
© 2021 Wiley-VCH GmbH.
Conflict of interest statement
Figures
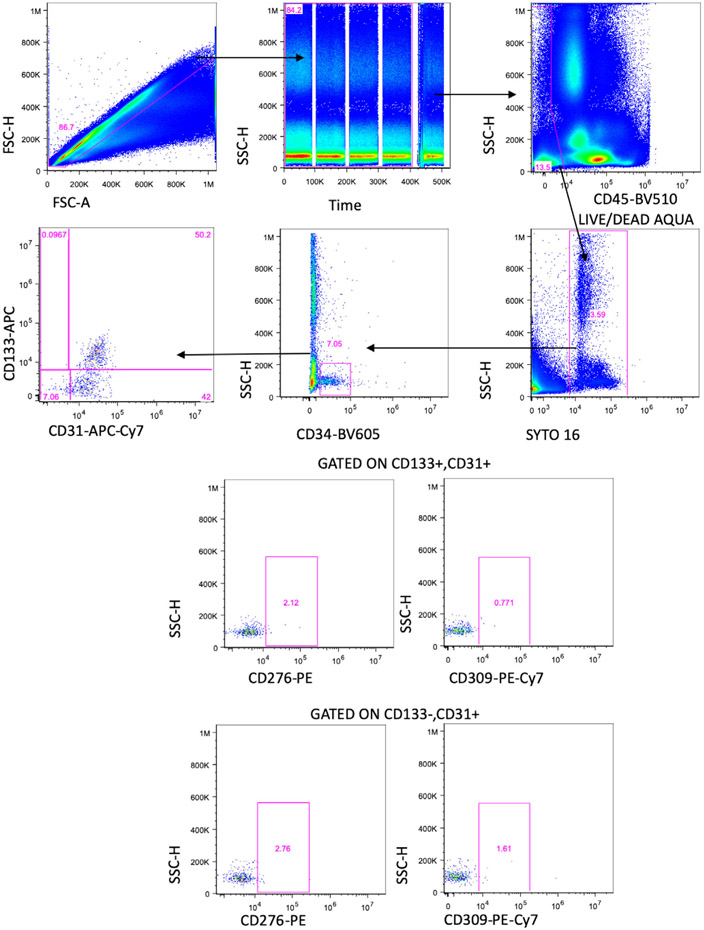
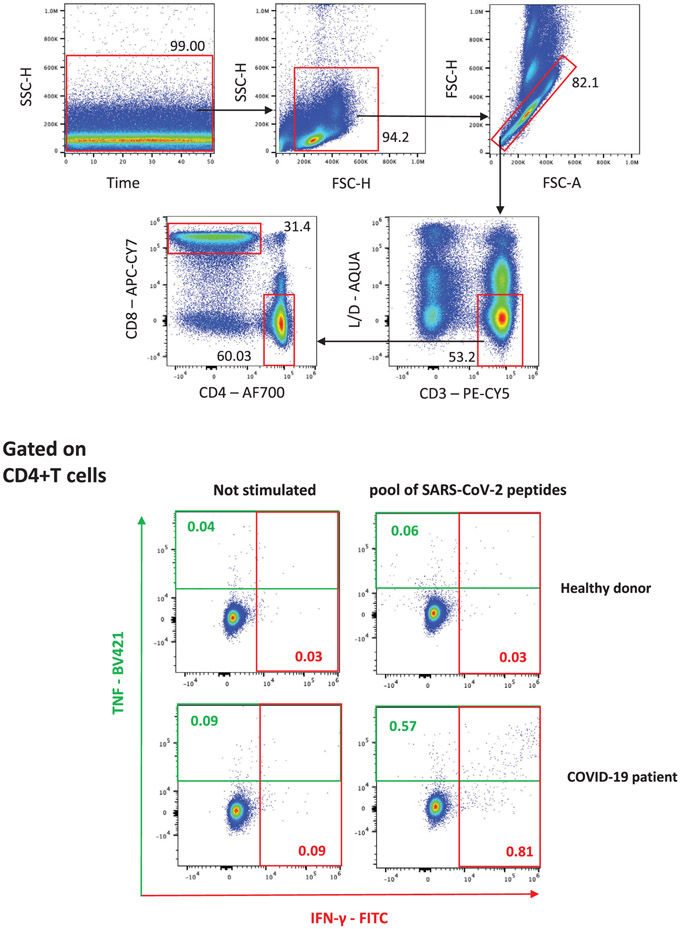
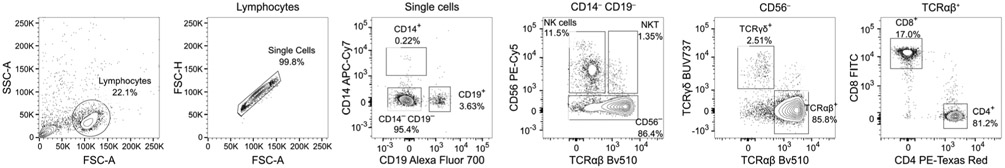
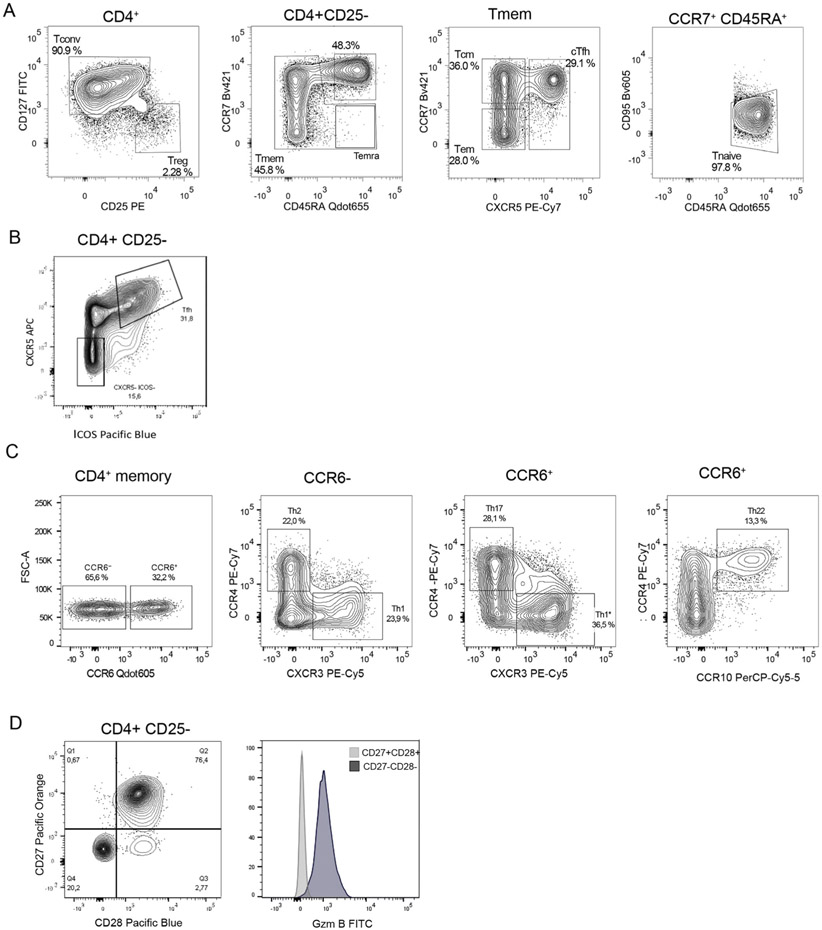
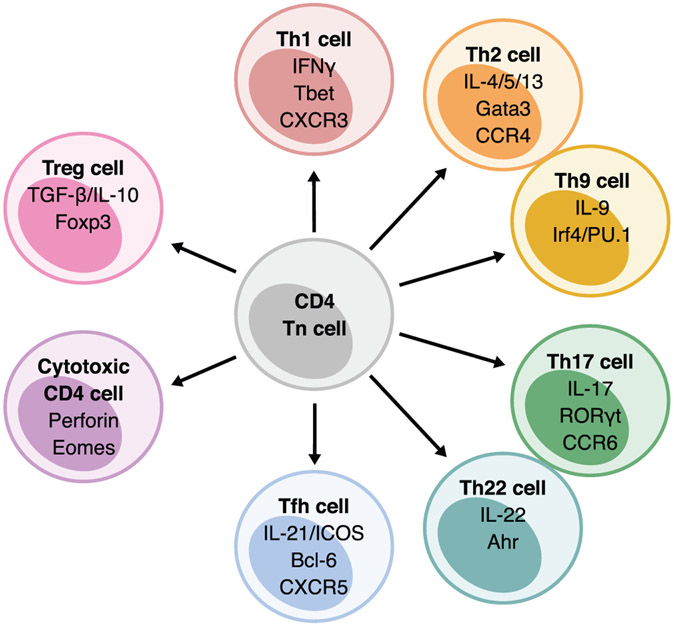
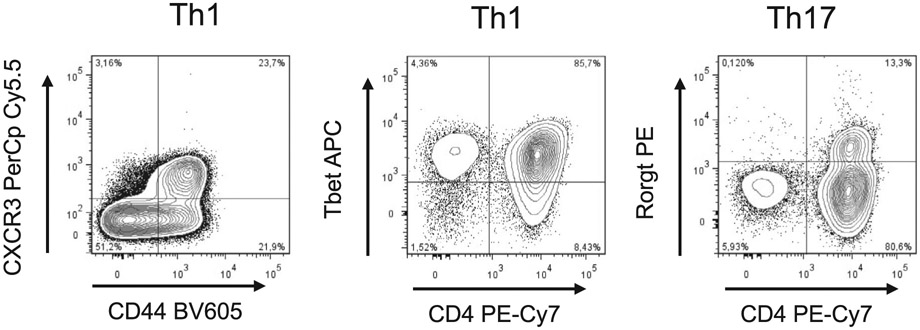
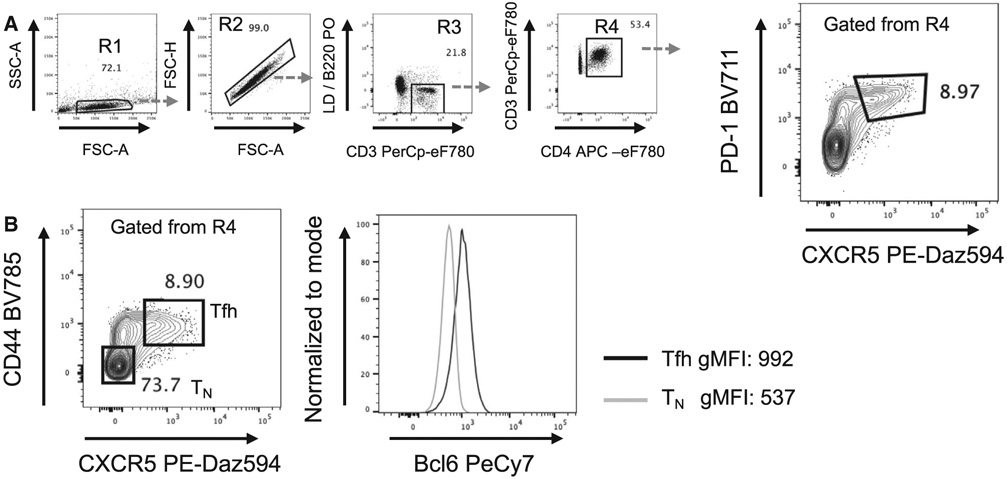
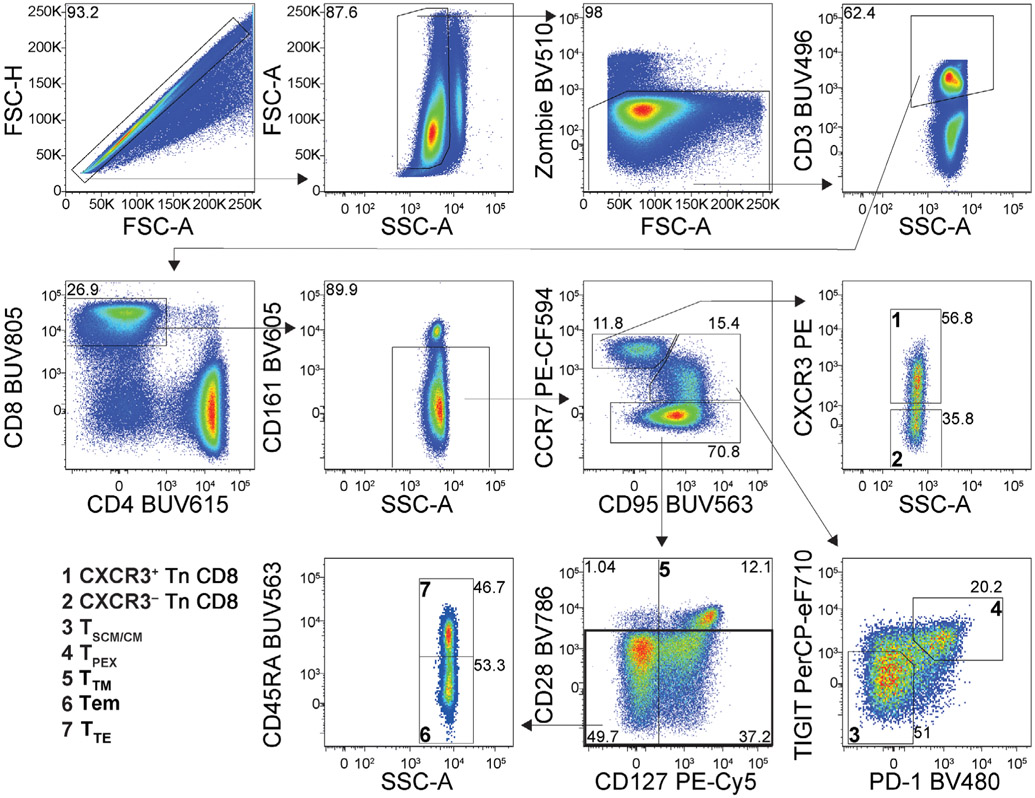
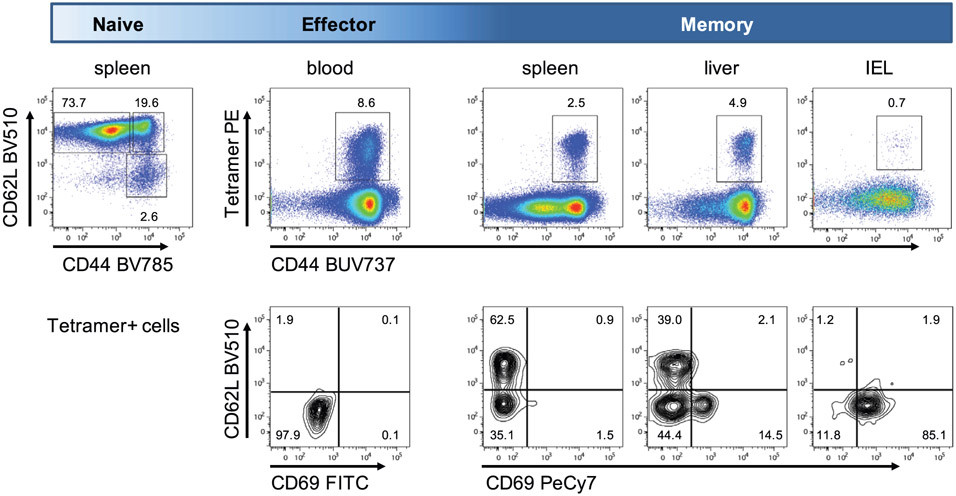
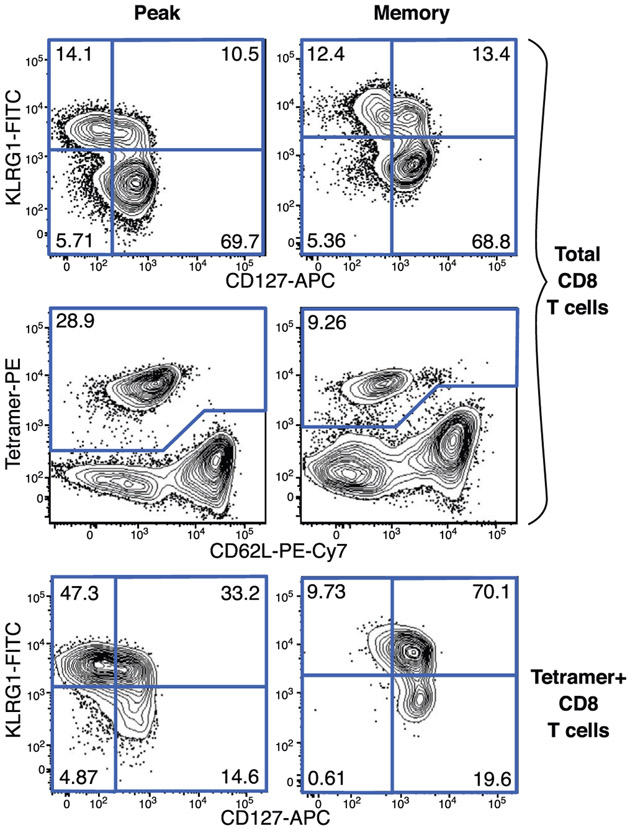
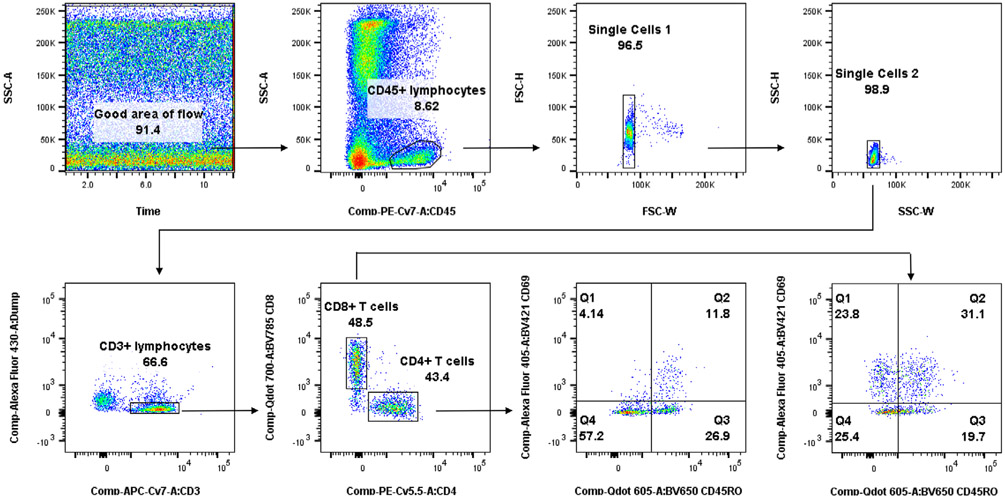
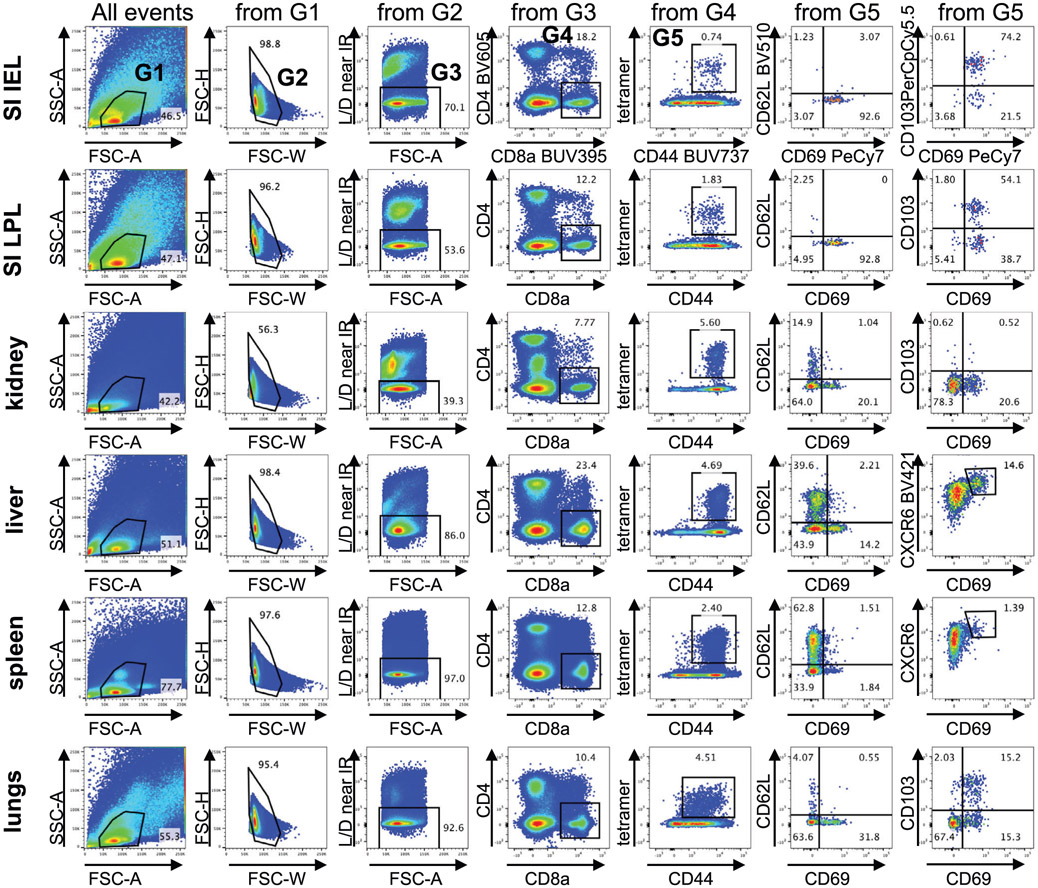
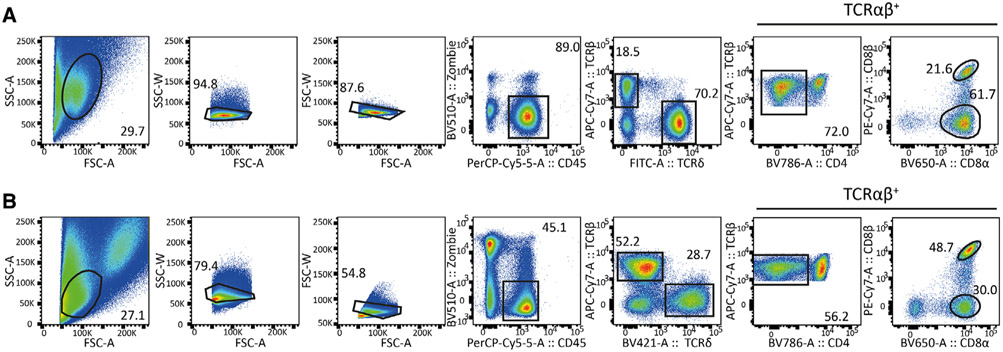
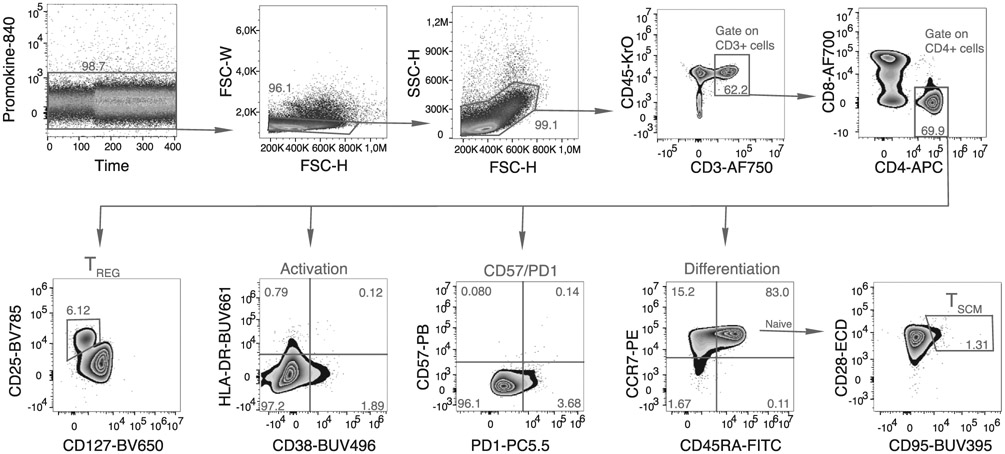
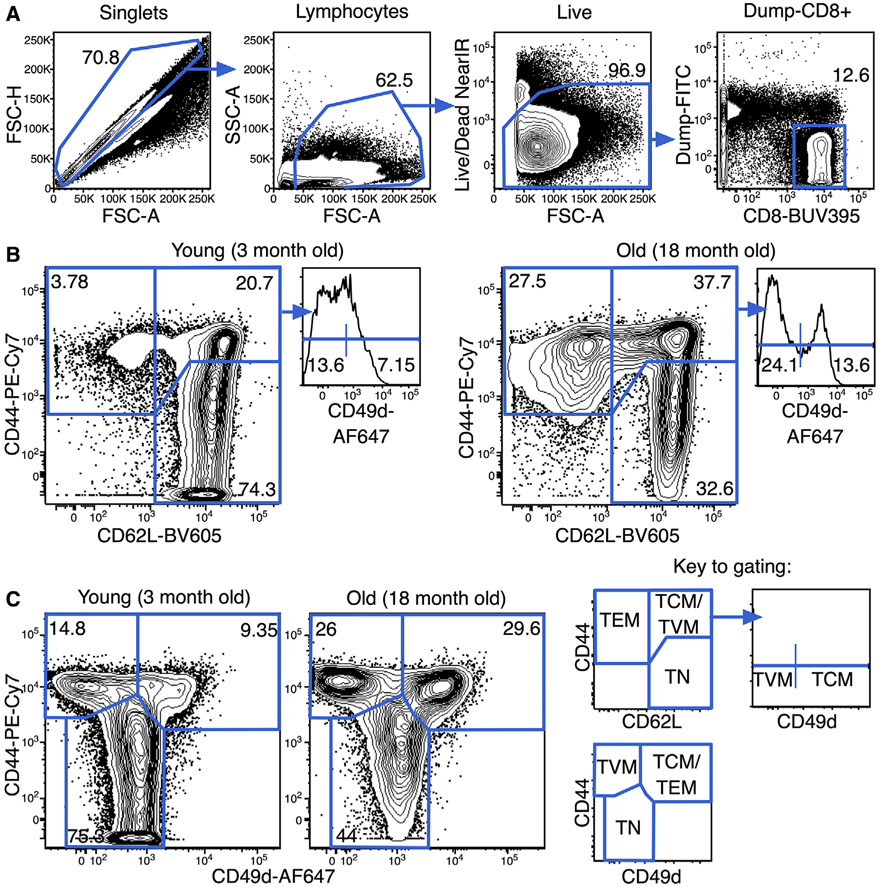
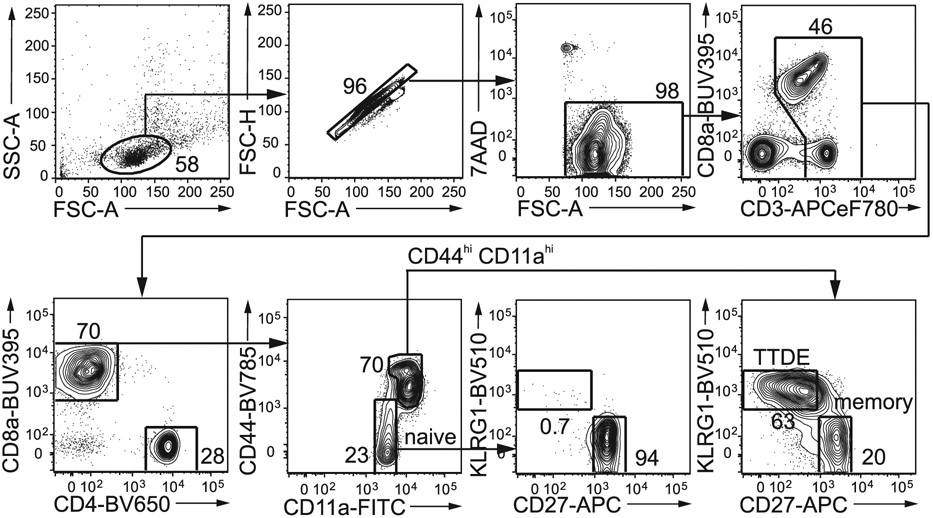
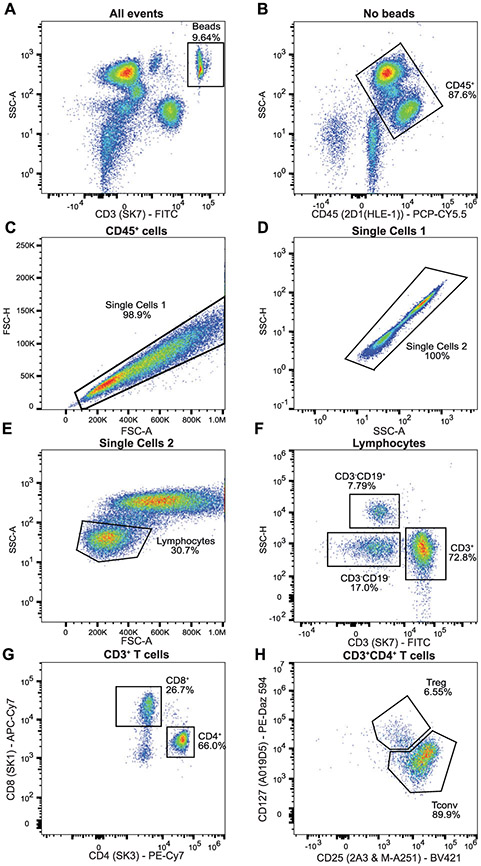
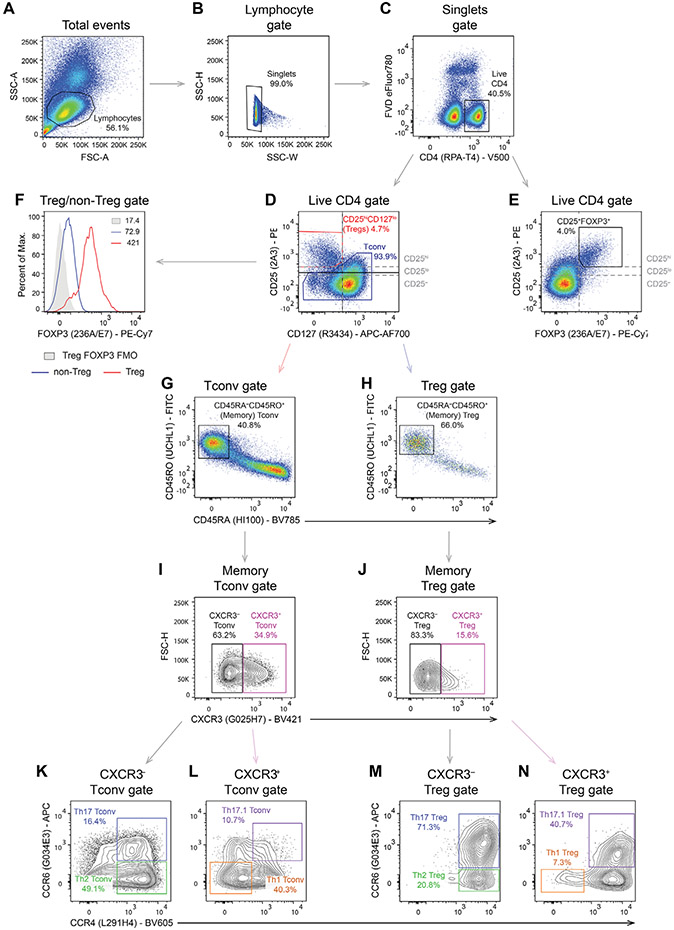
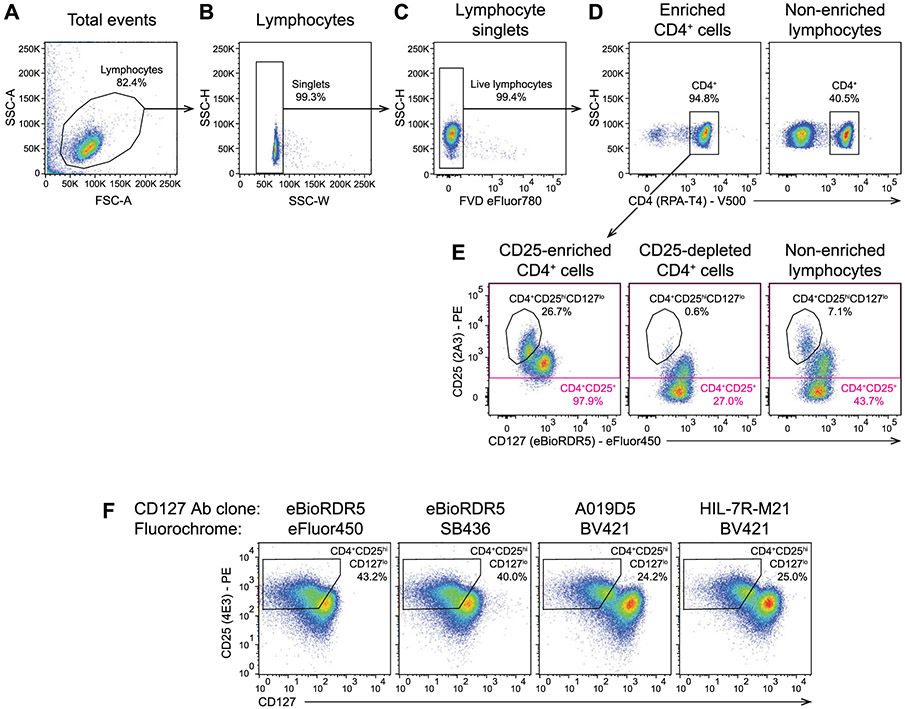
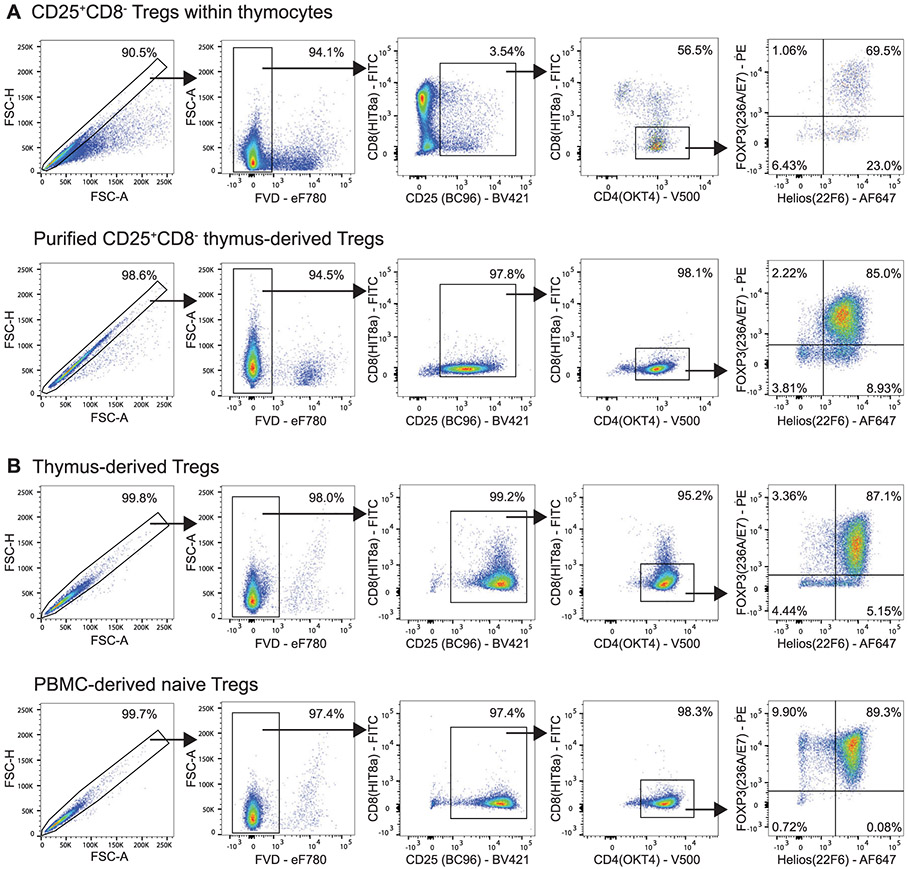
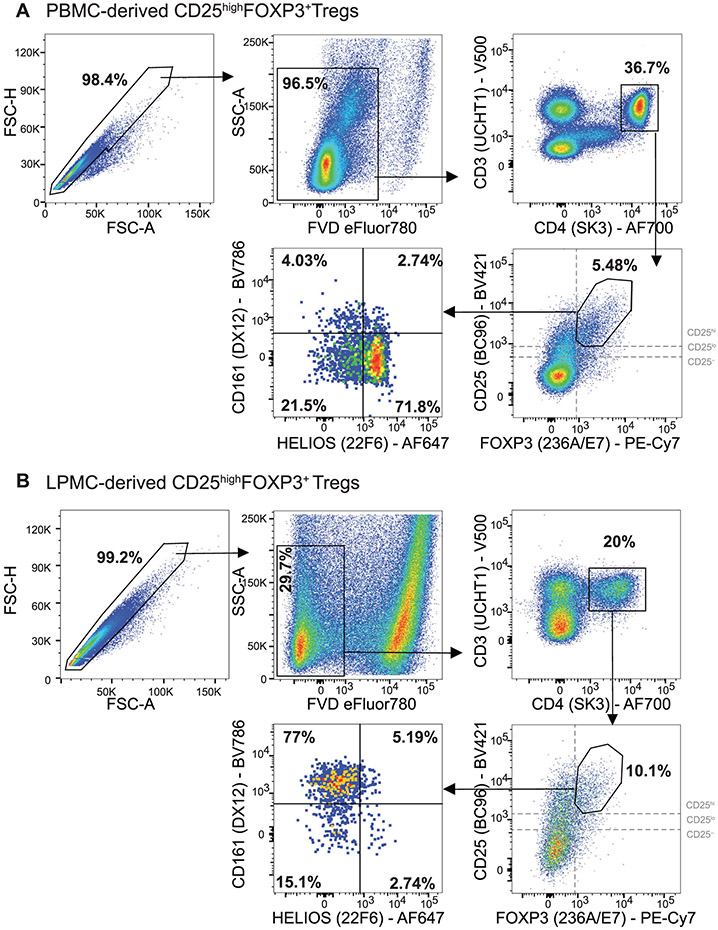
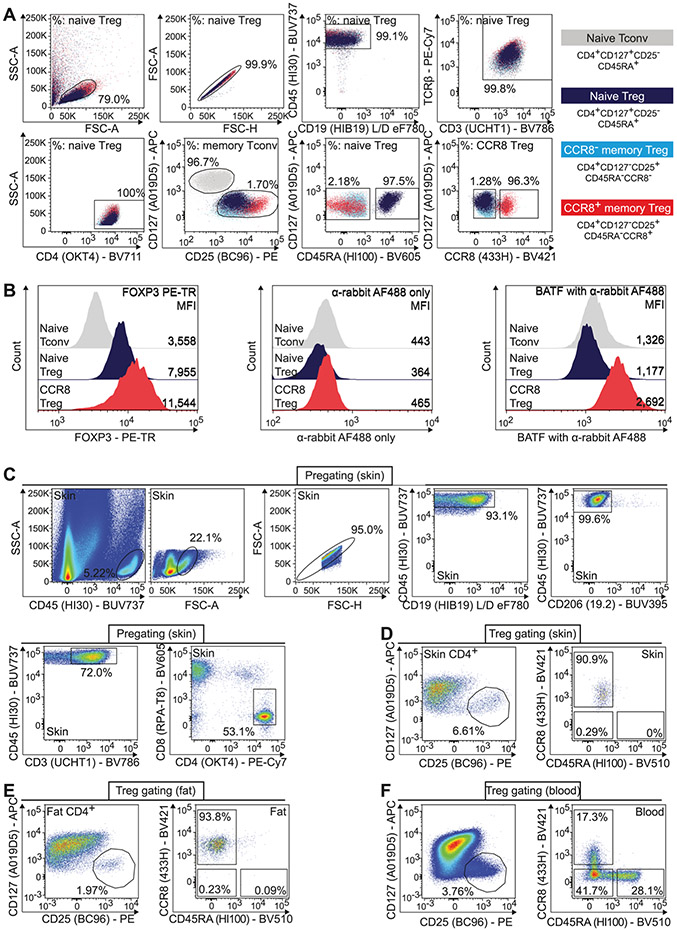
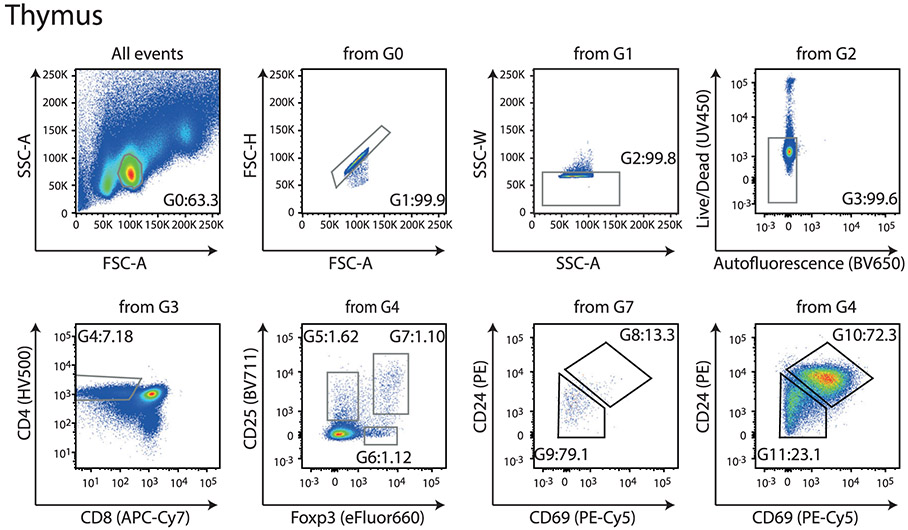
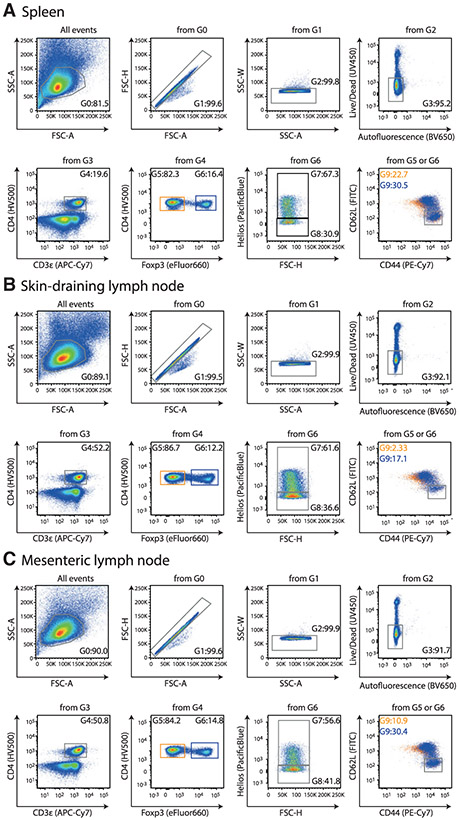
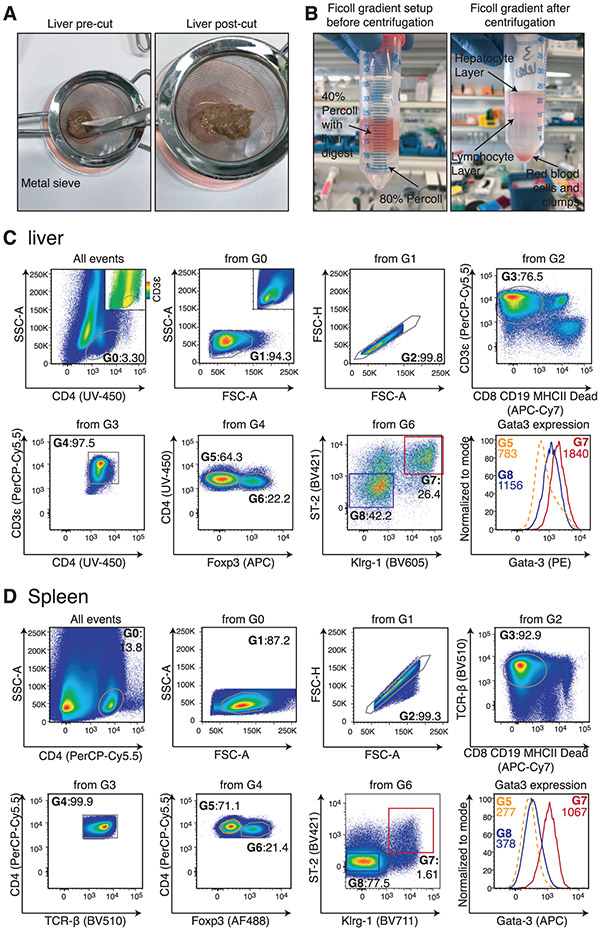
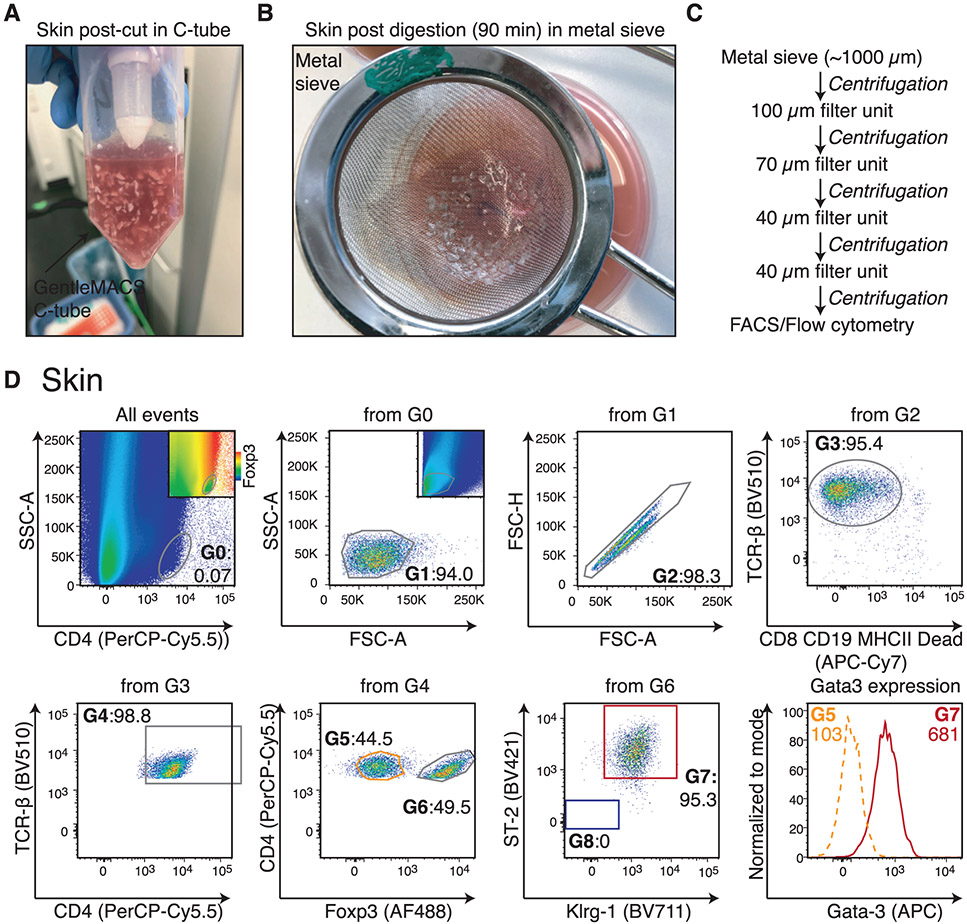
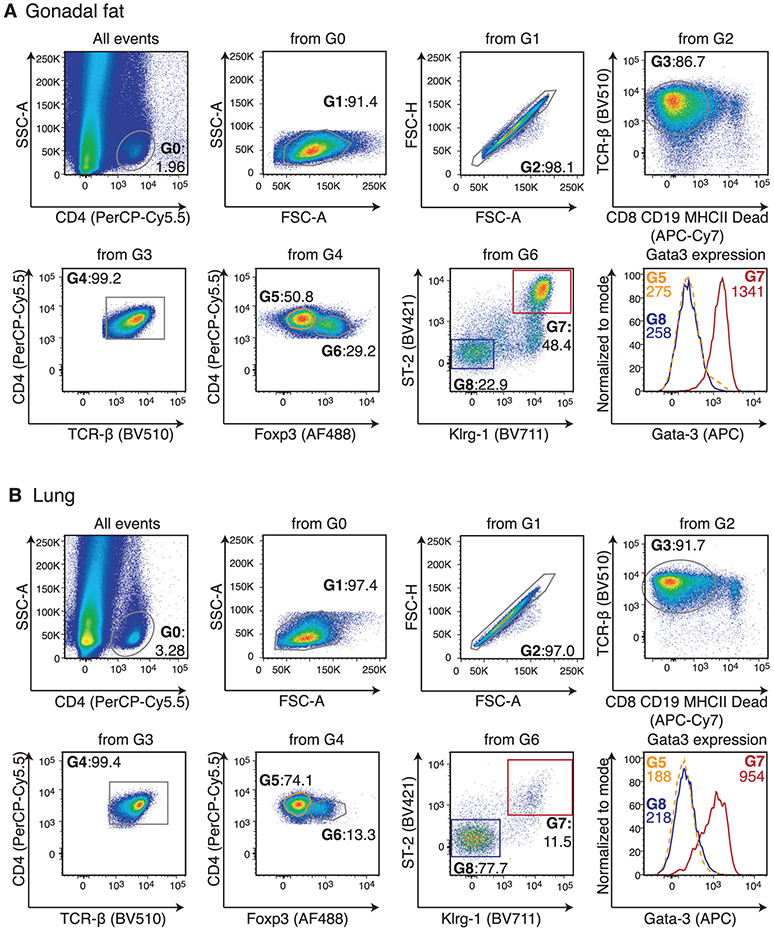
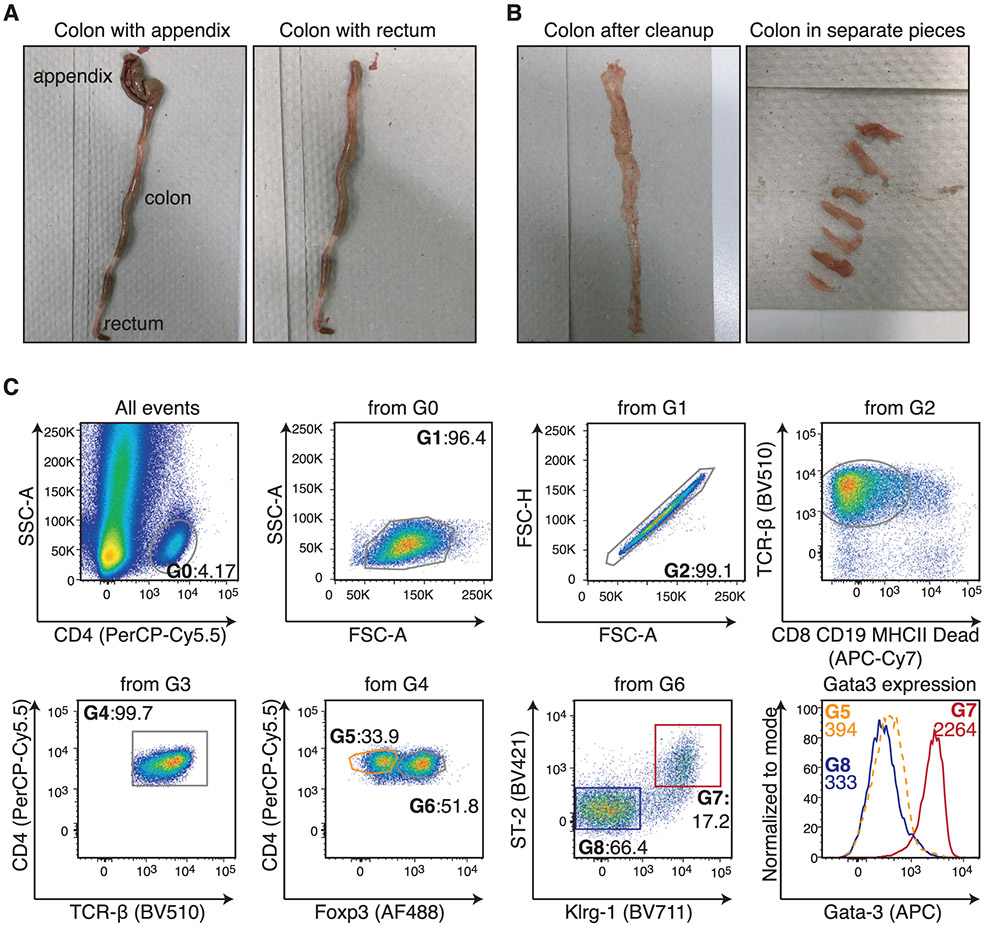
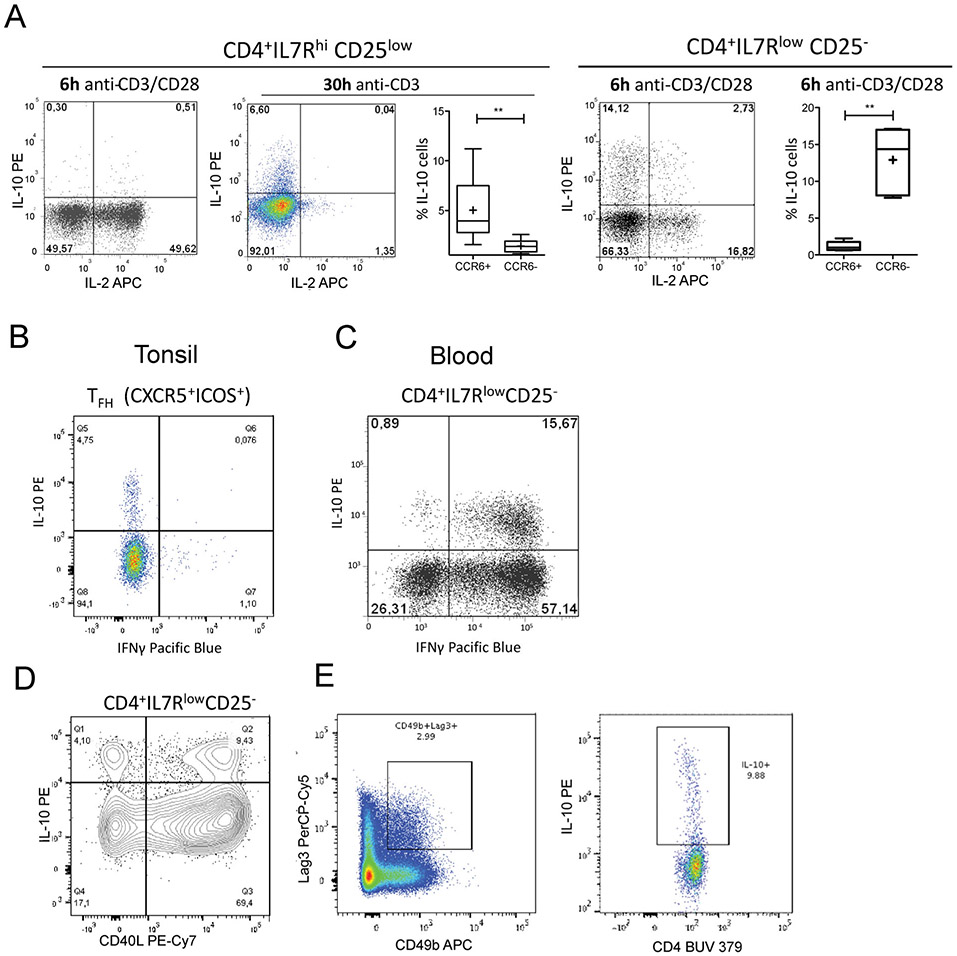
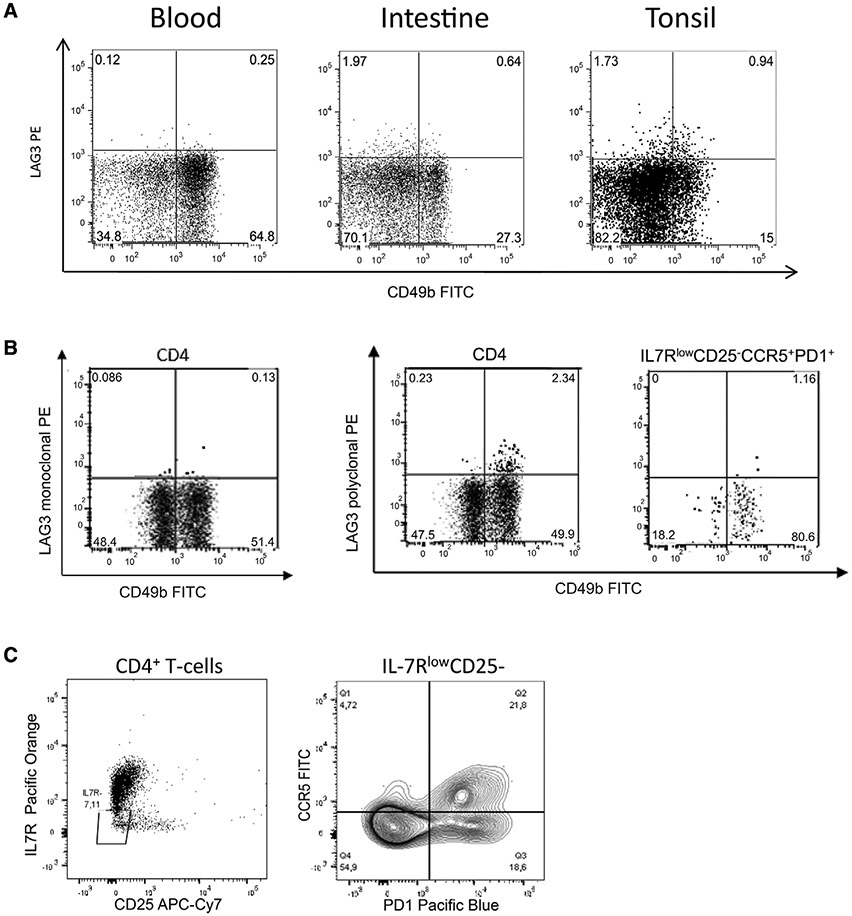
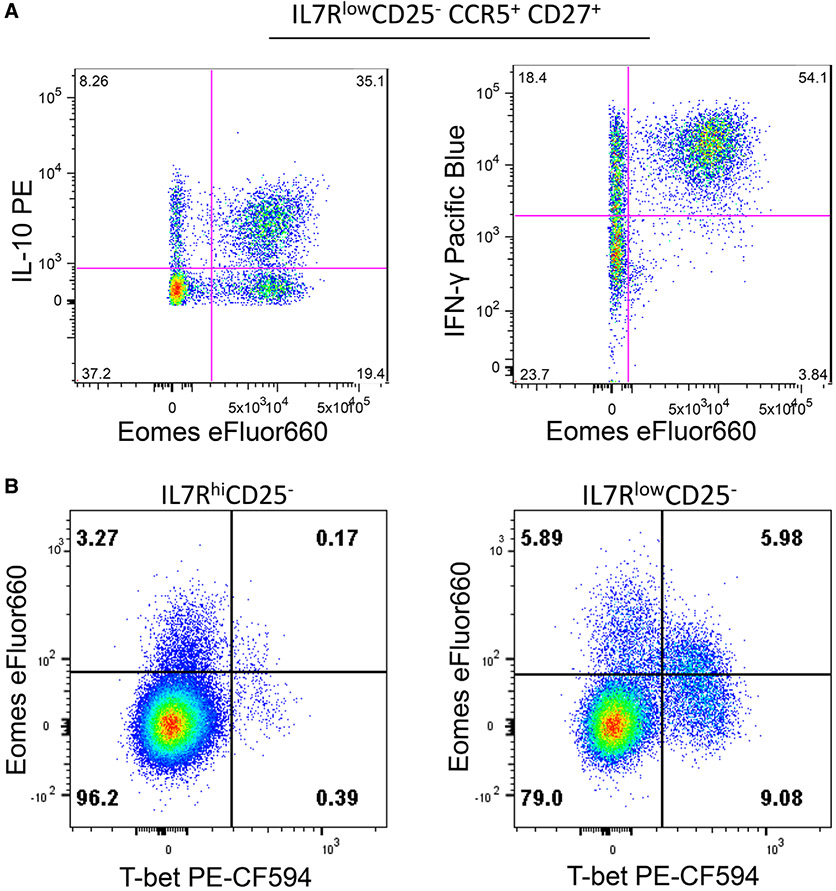
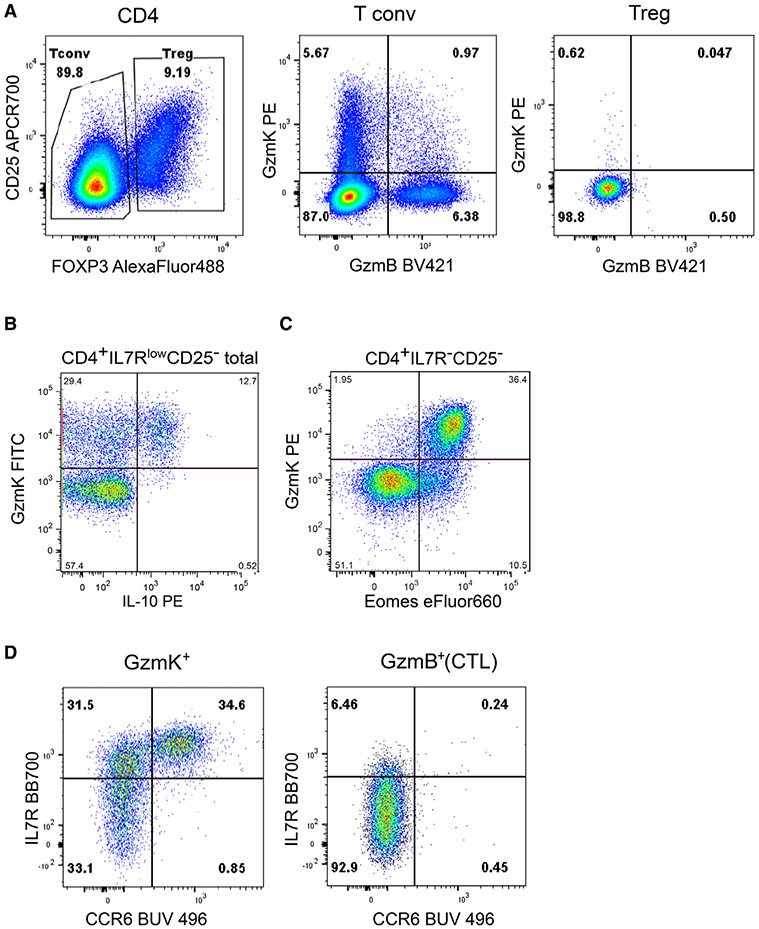
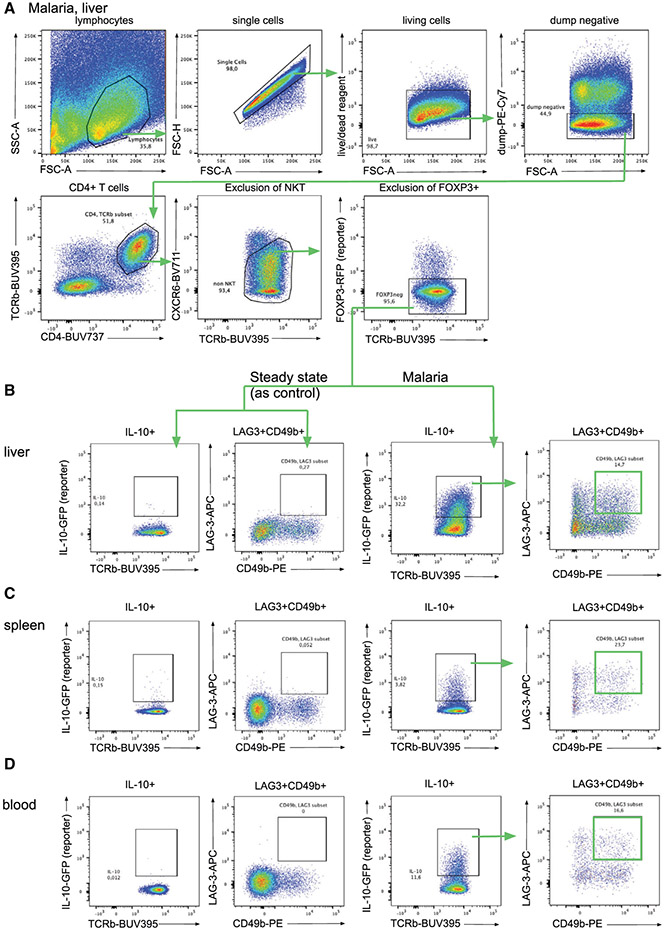
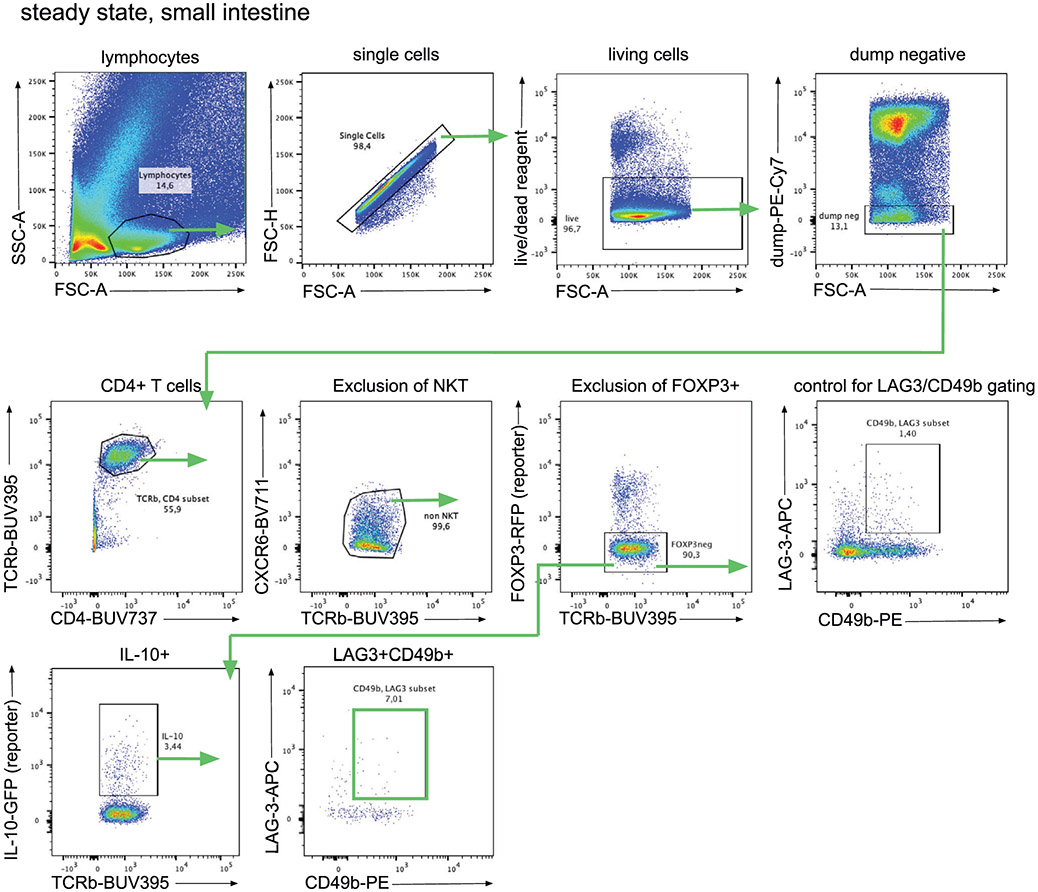
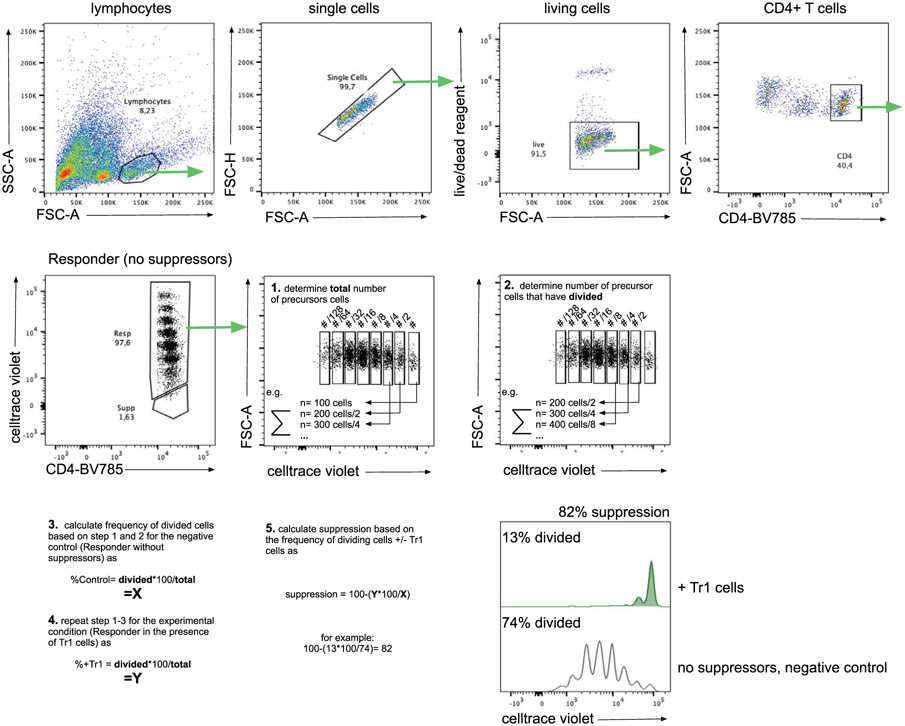
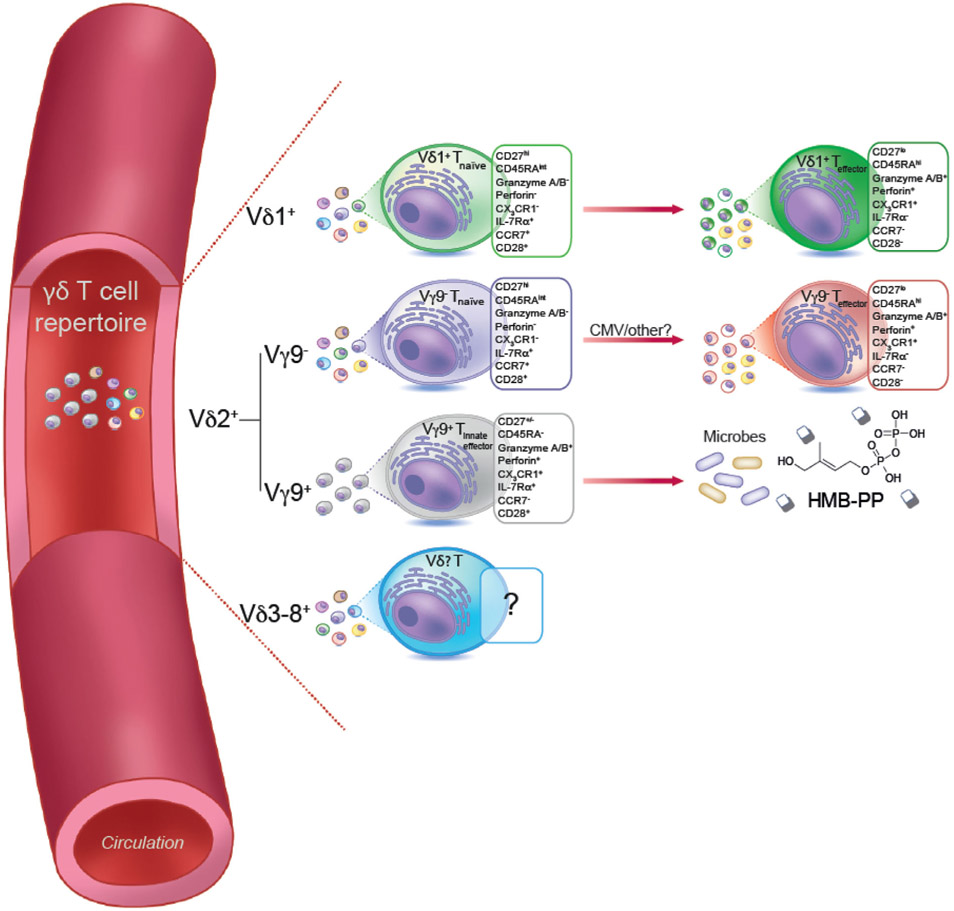
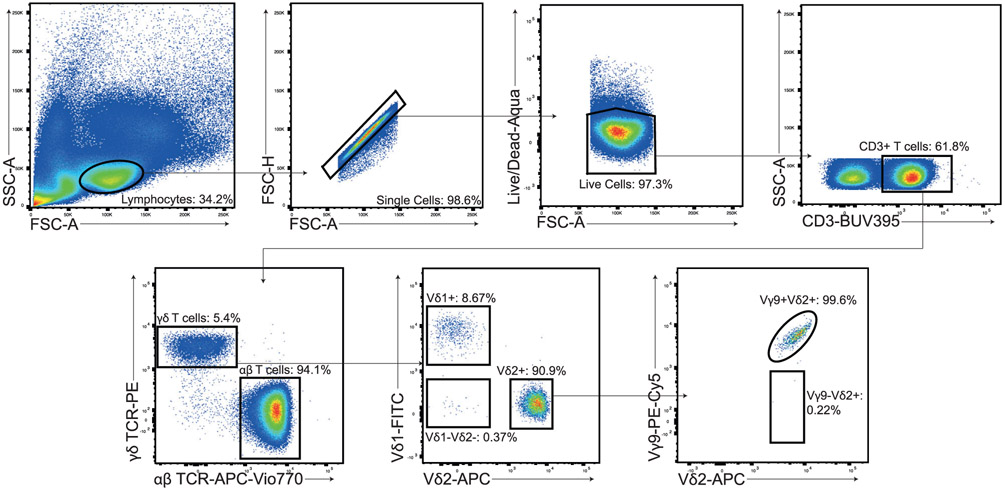
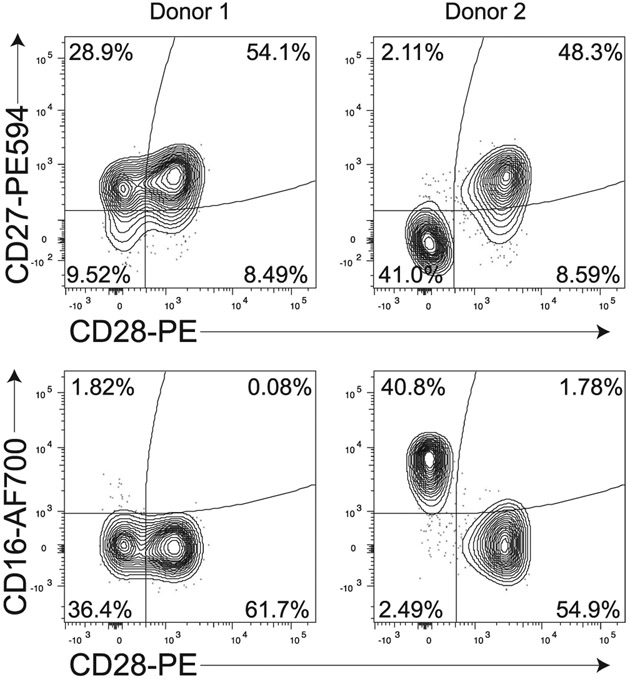
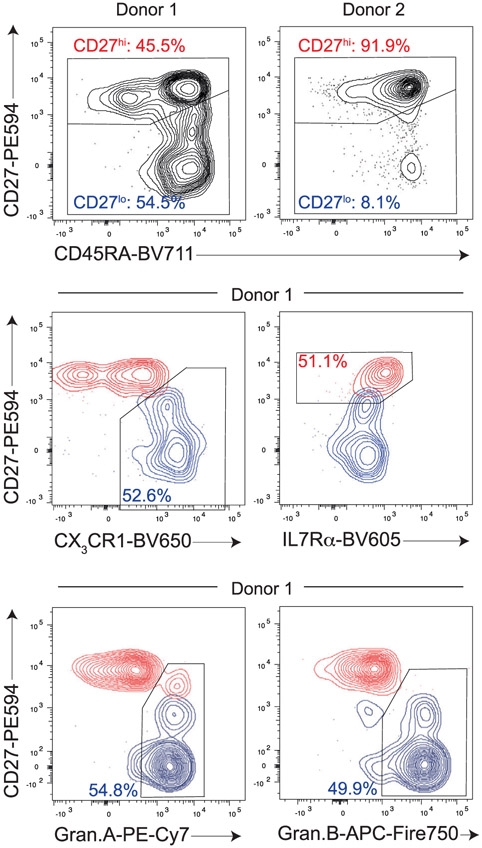
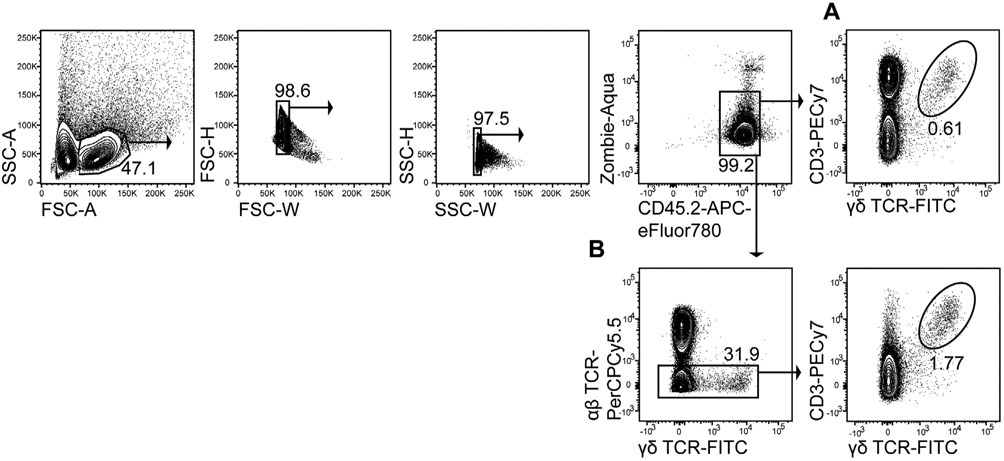
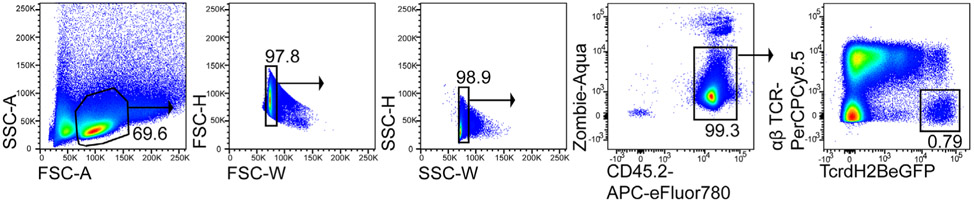
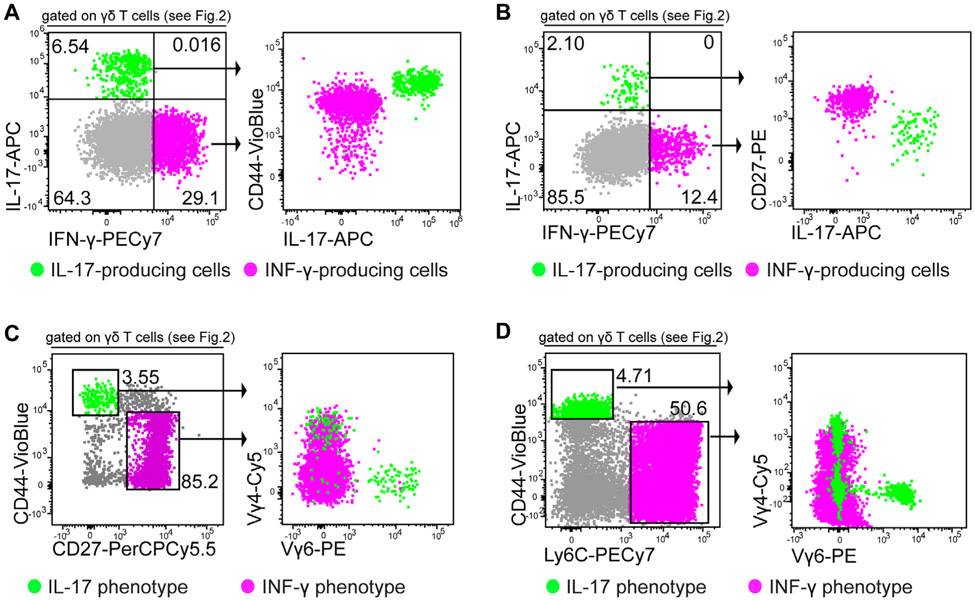
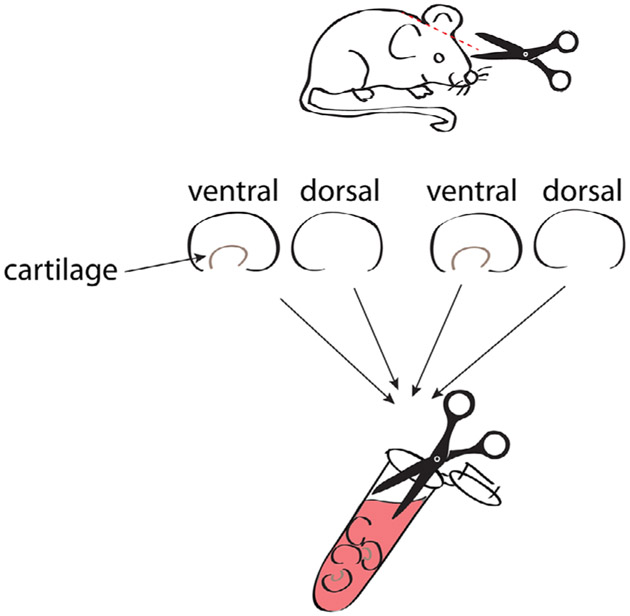
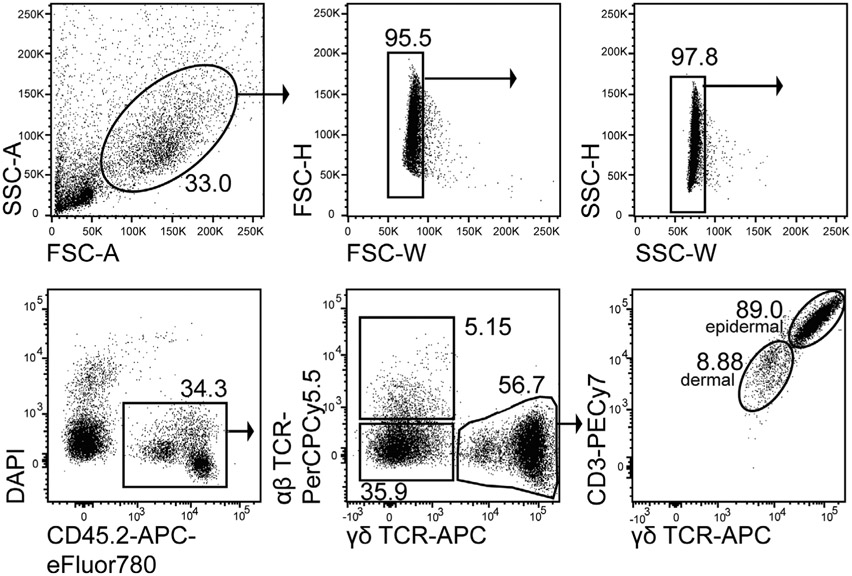
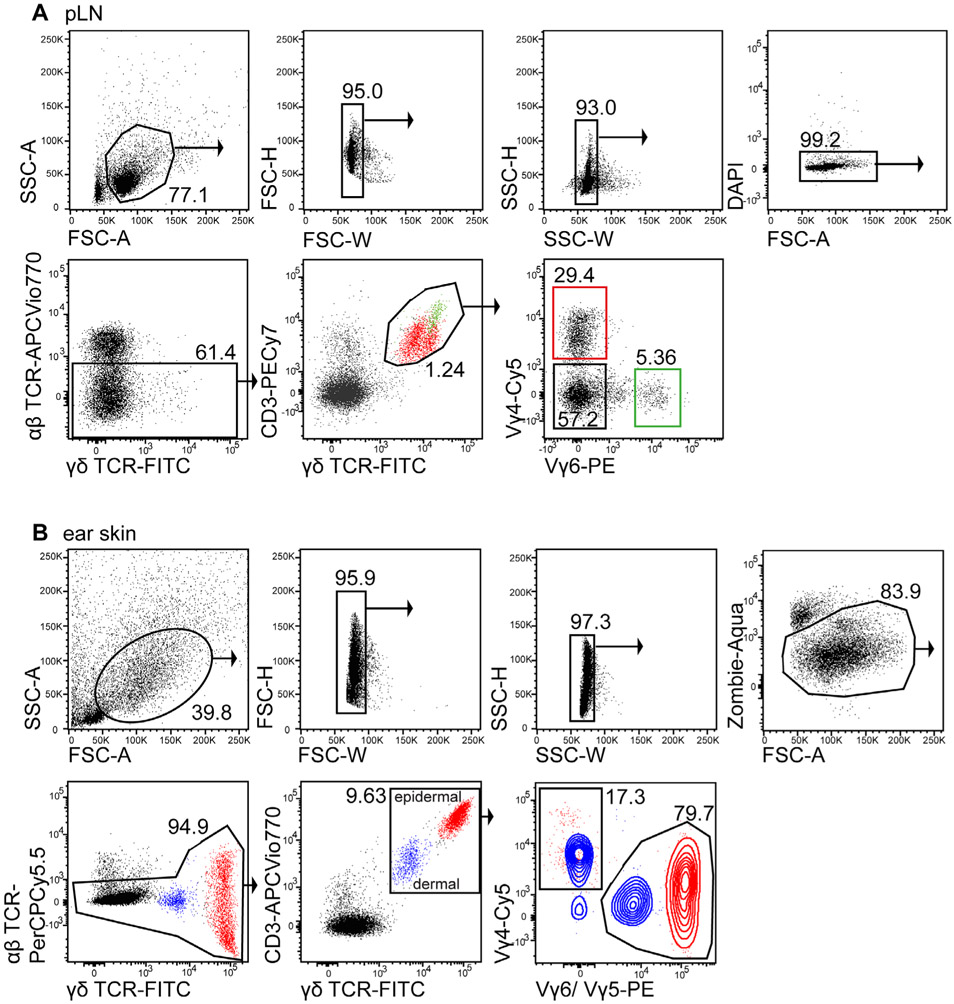
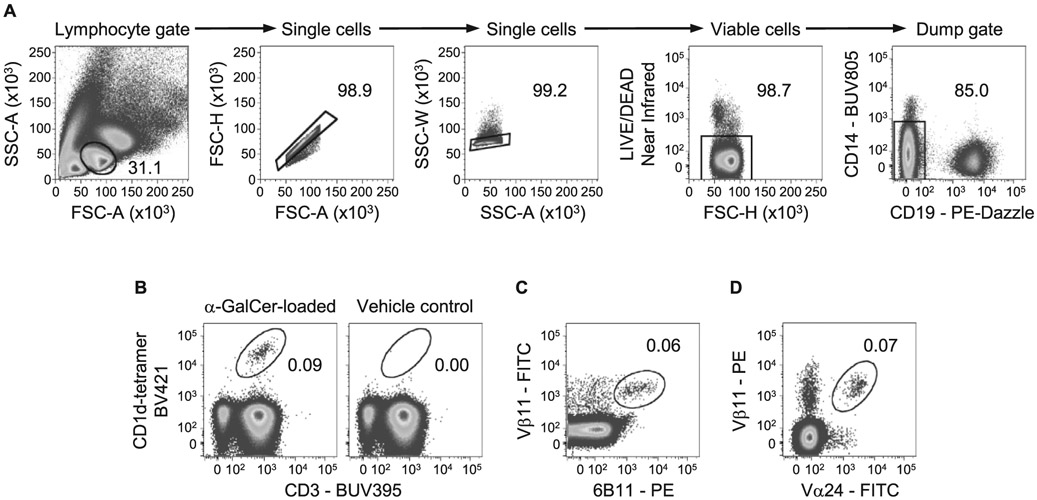
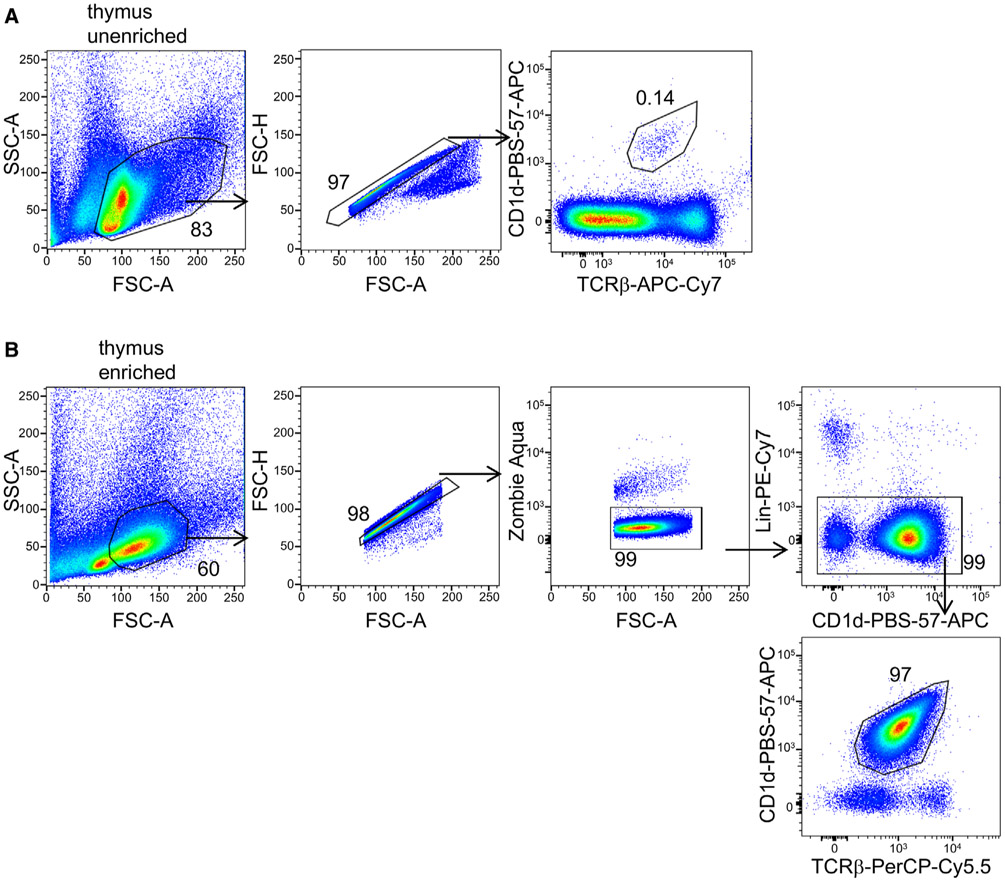
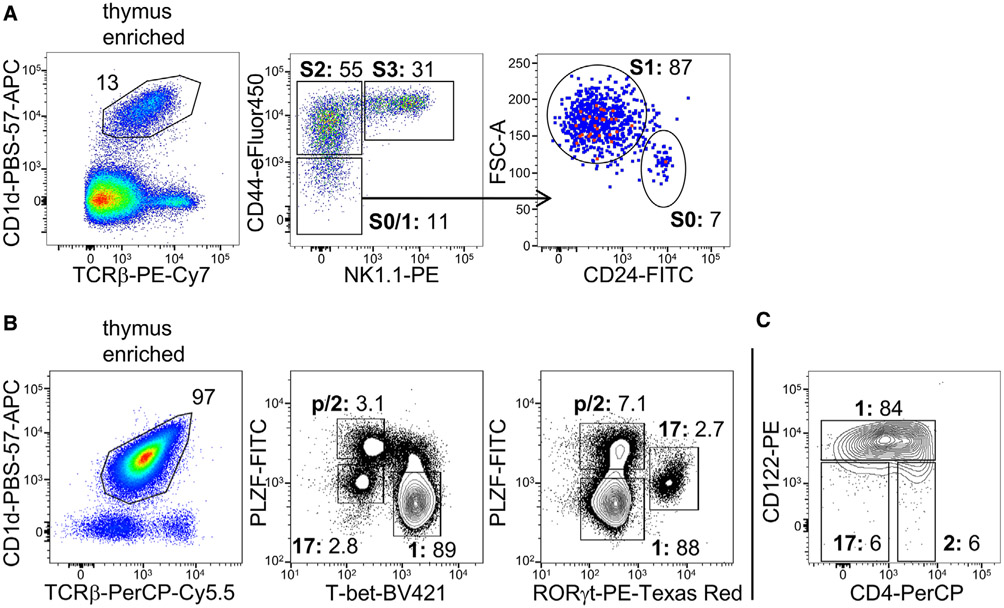
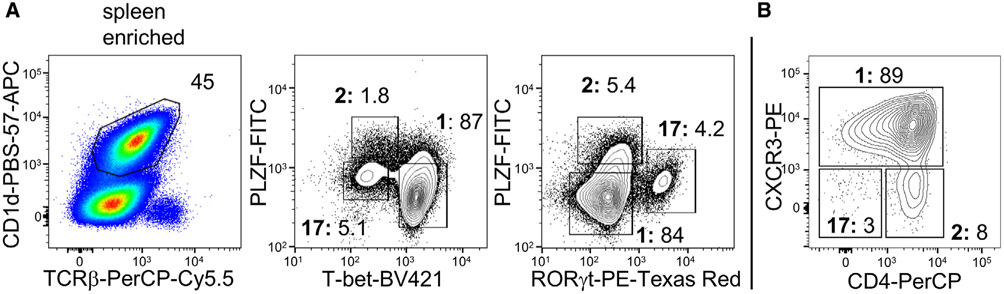
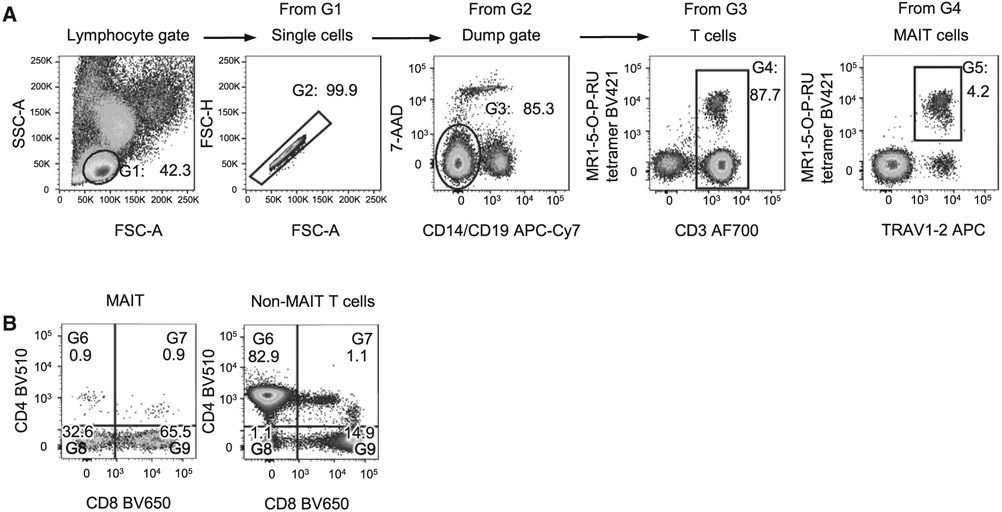
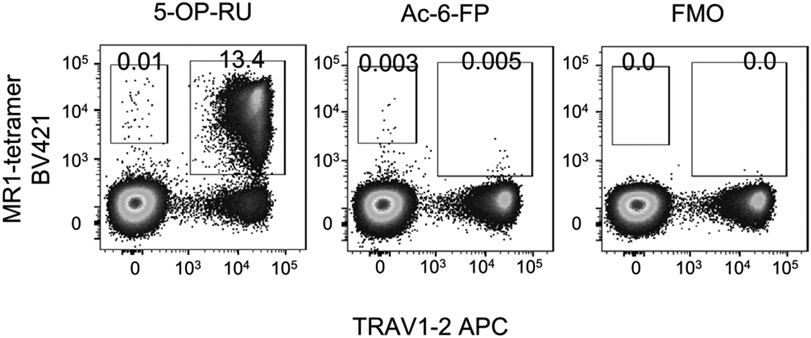
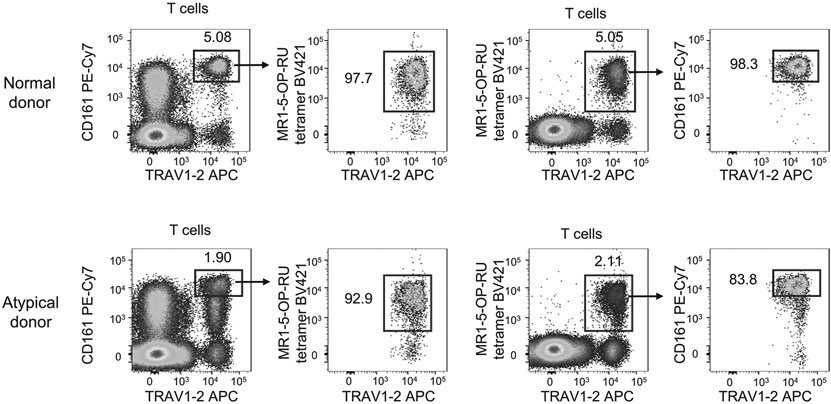
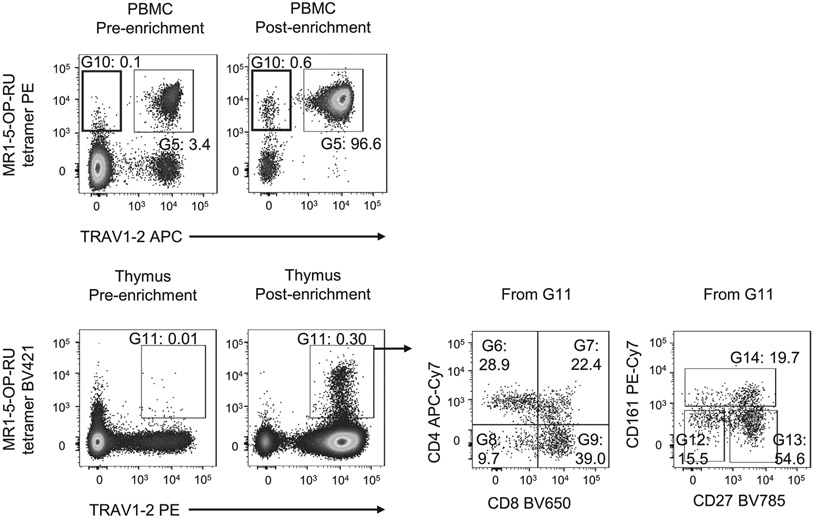
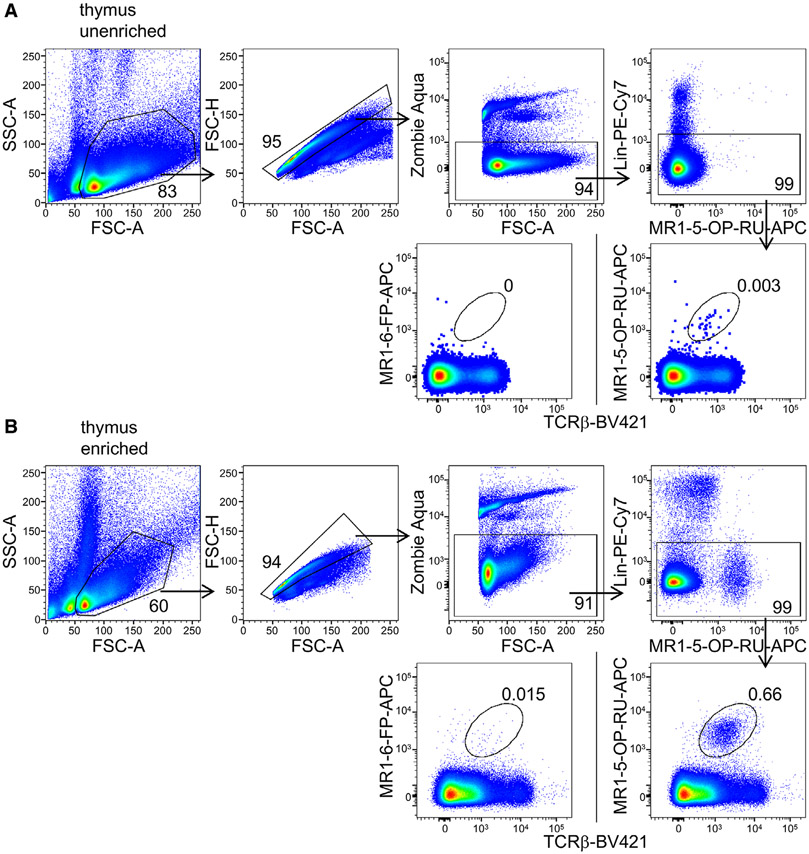
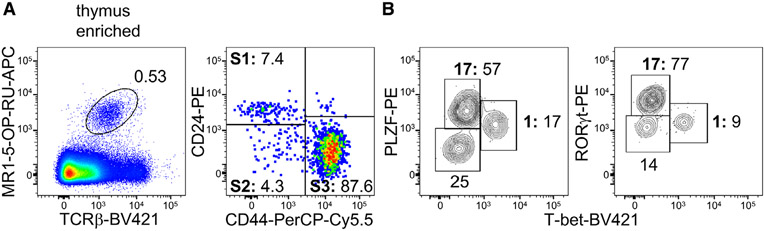
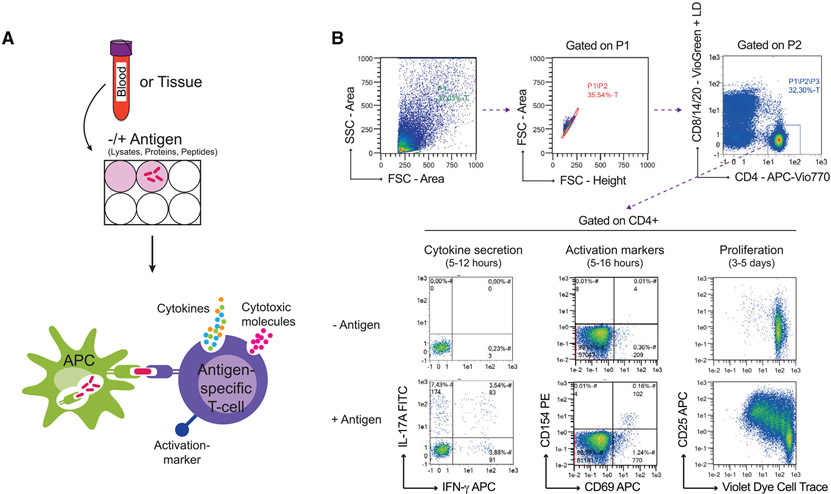
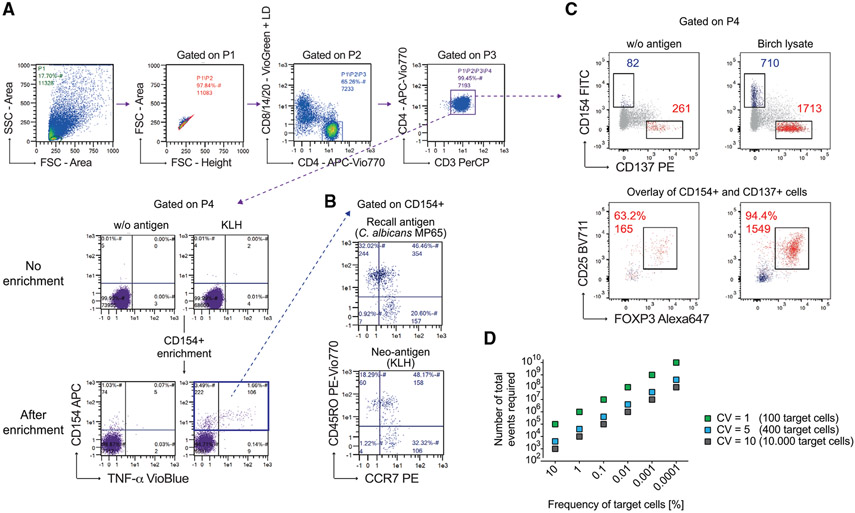
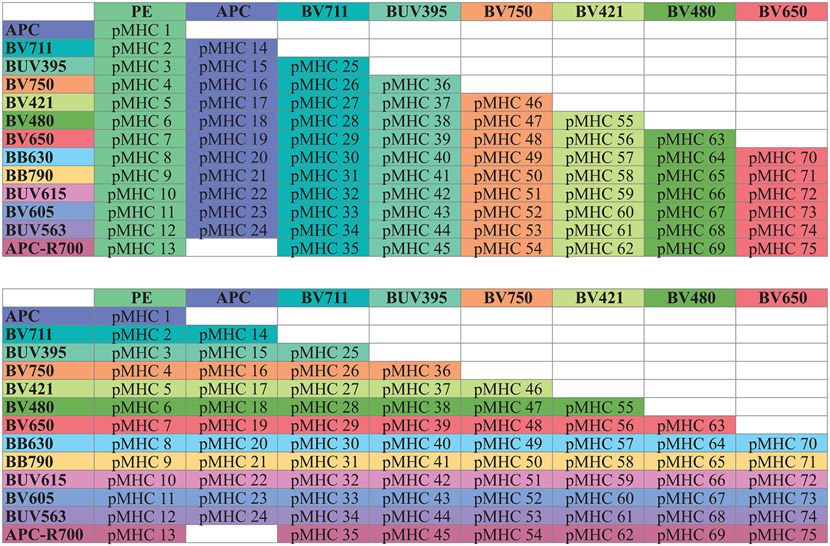
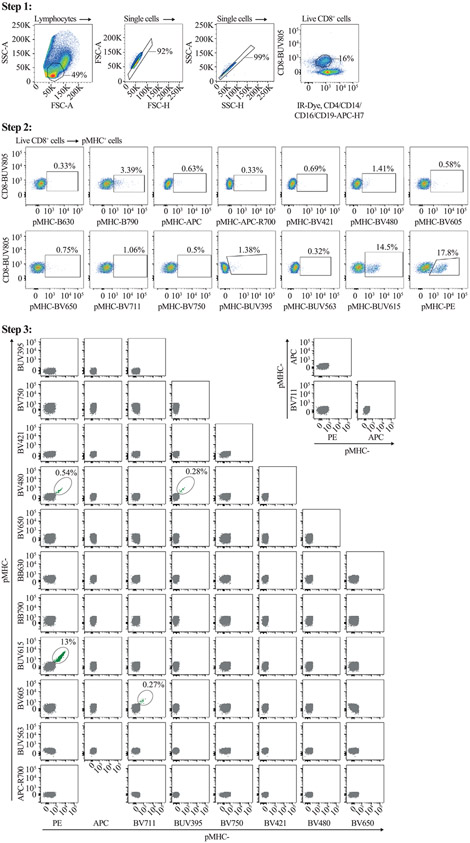
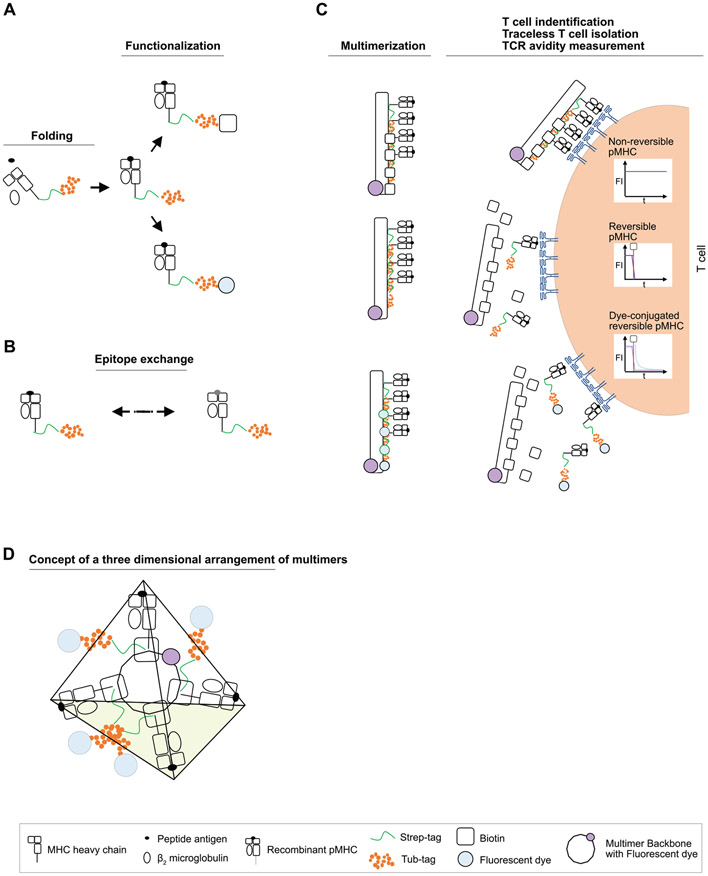
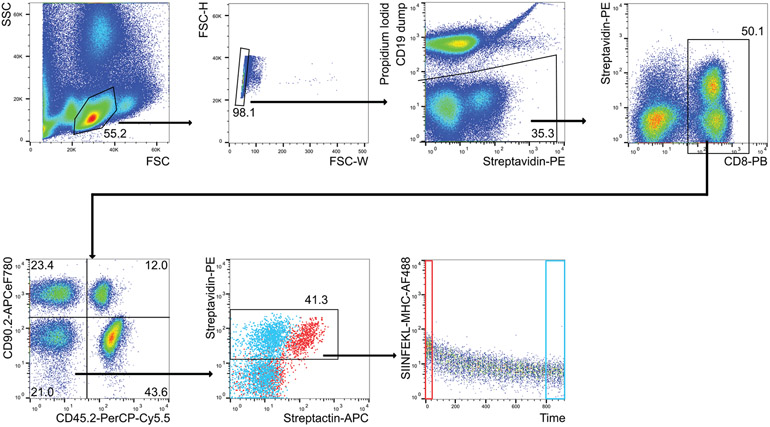
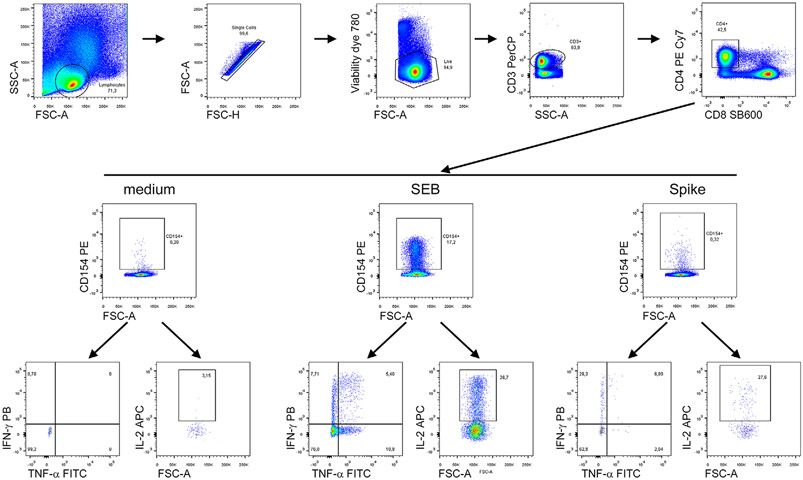
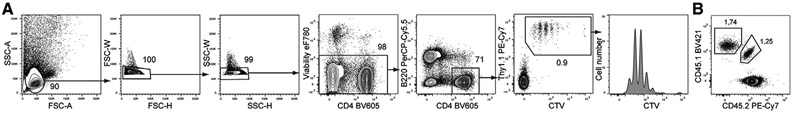
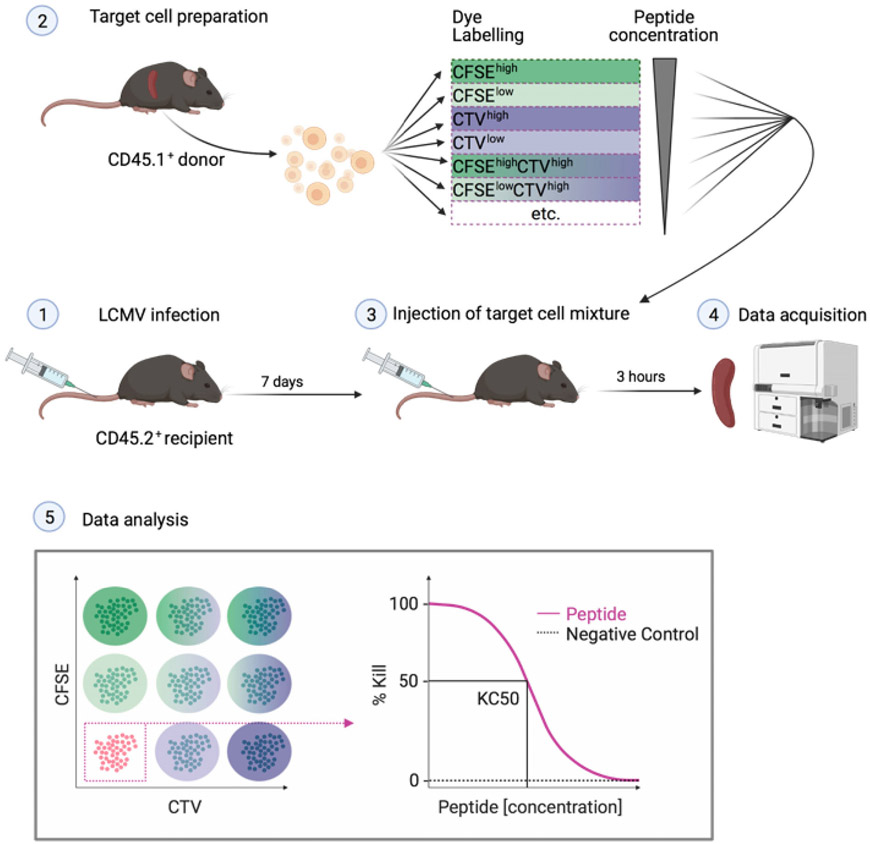
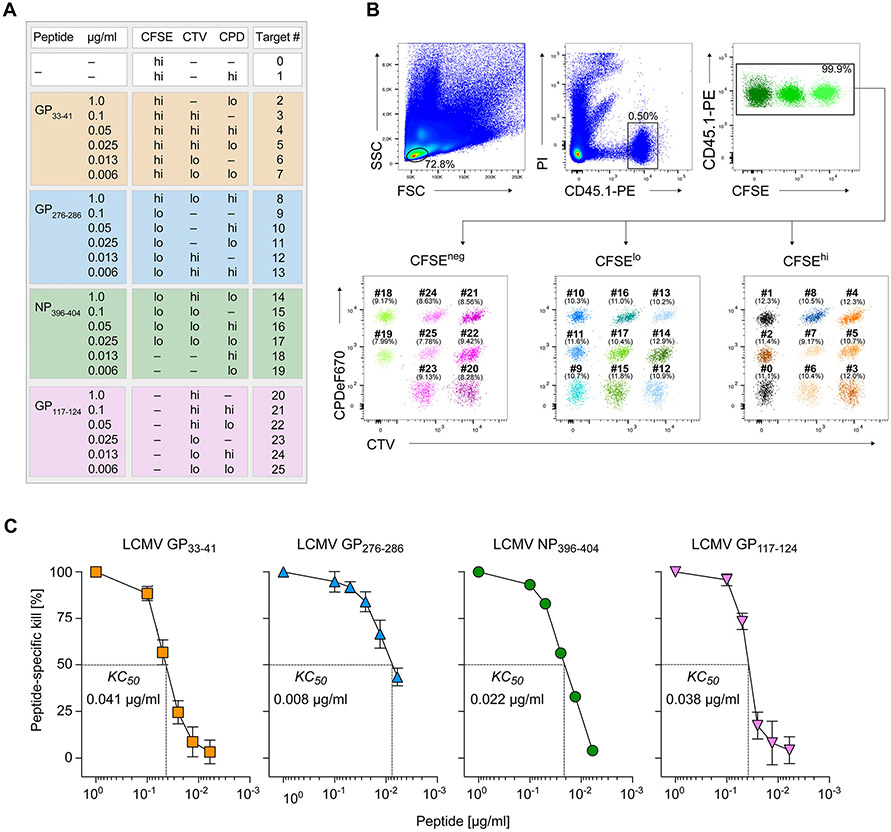
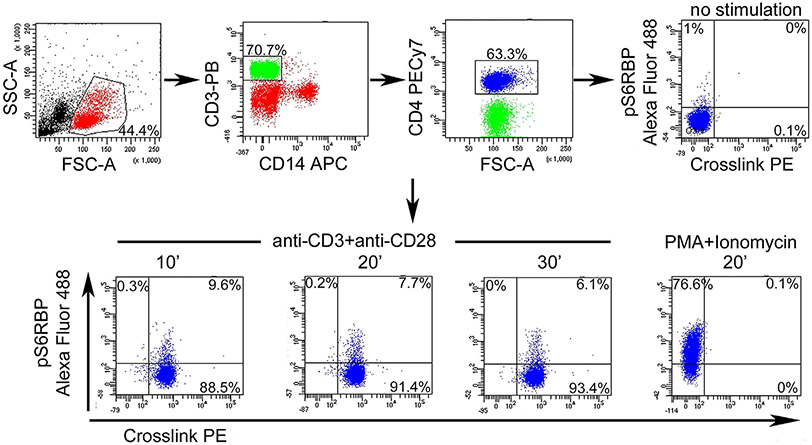
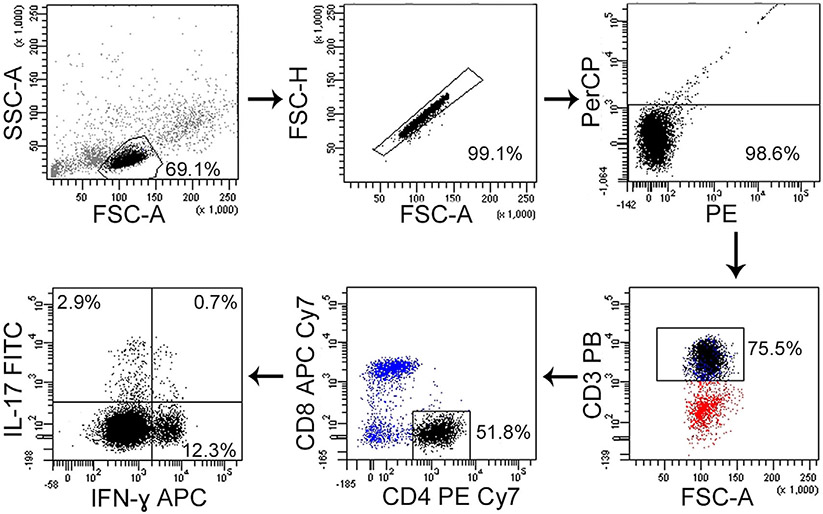
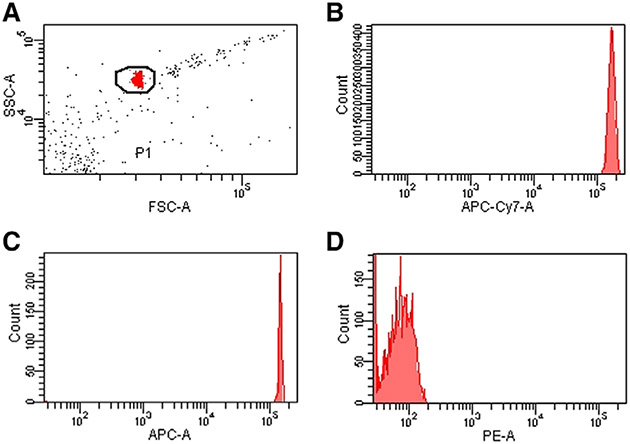
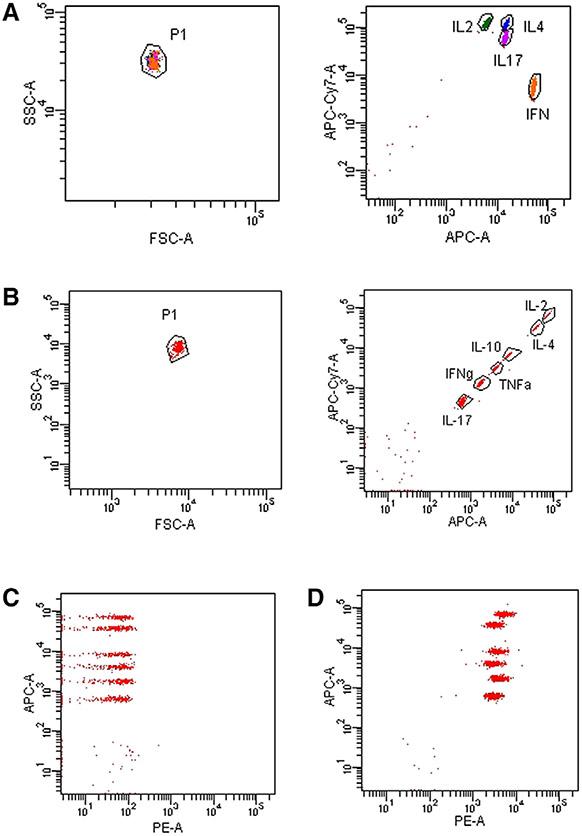
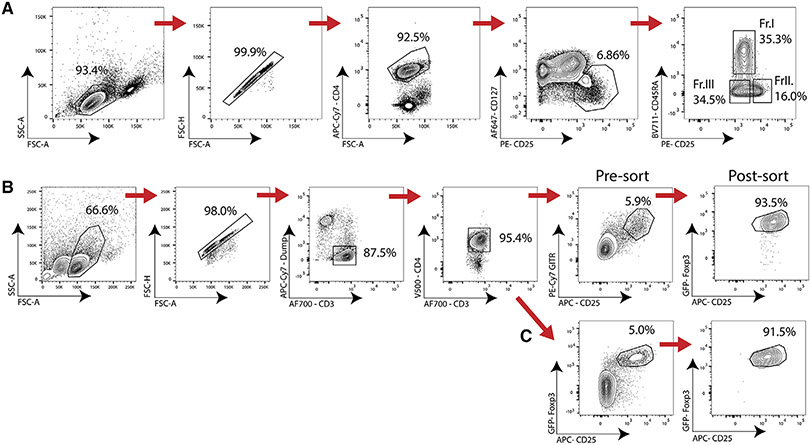
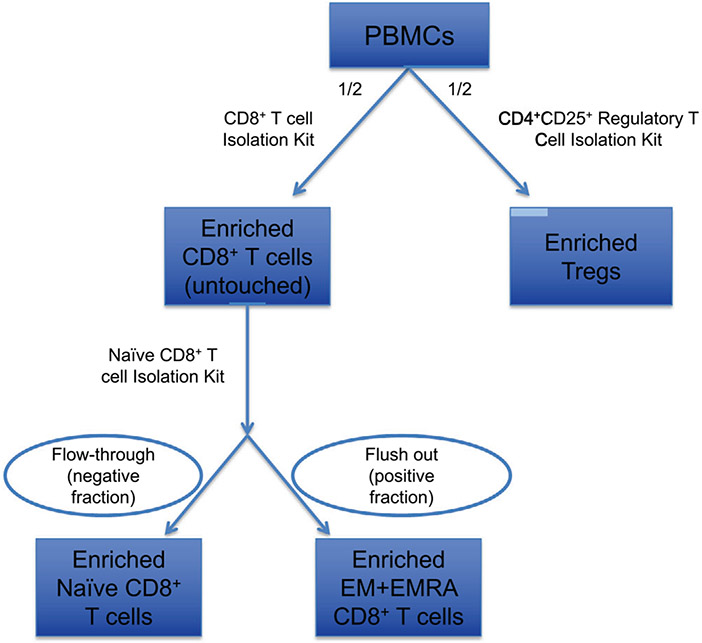
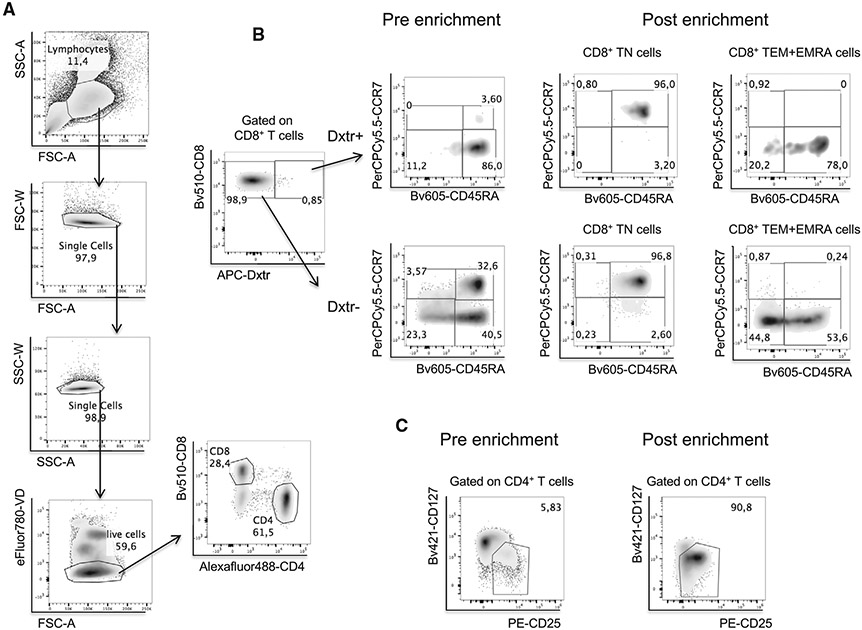
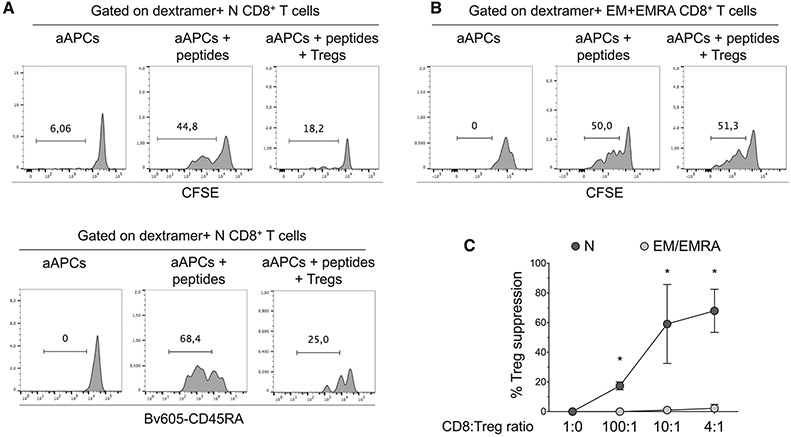
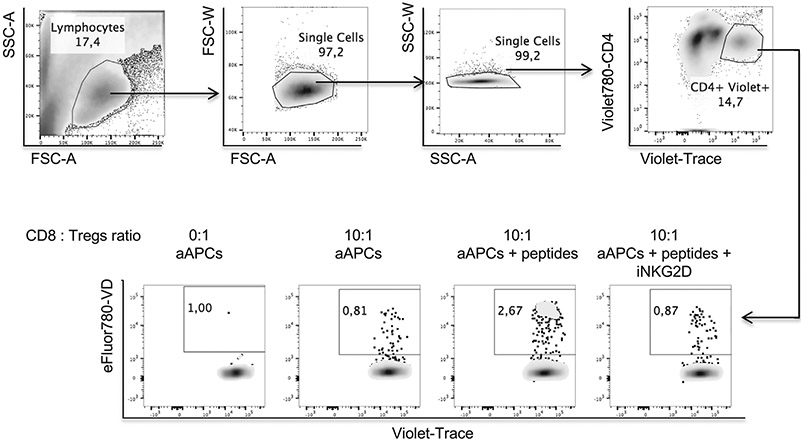
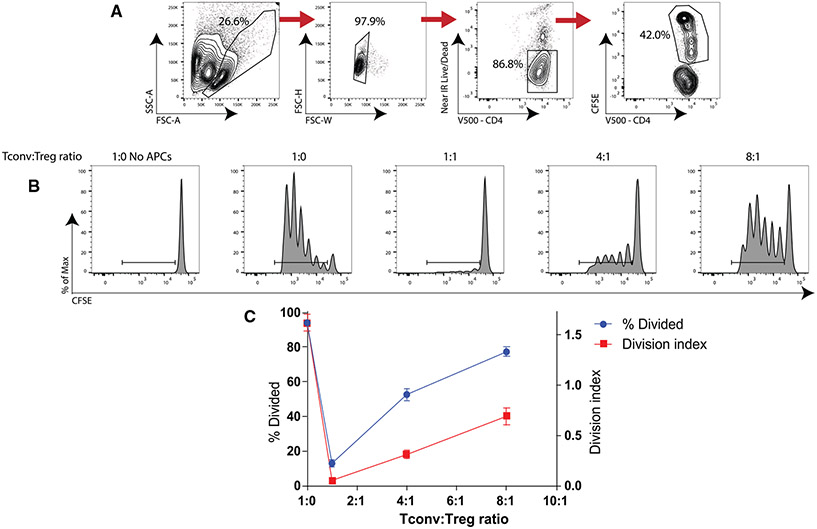
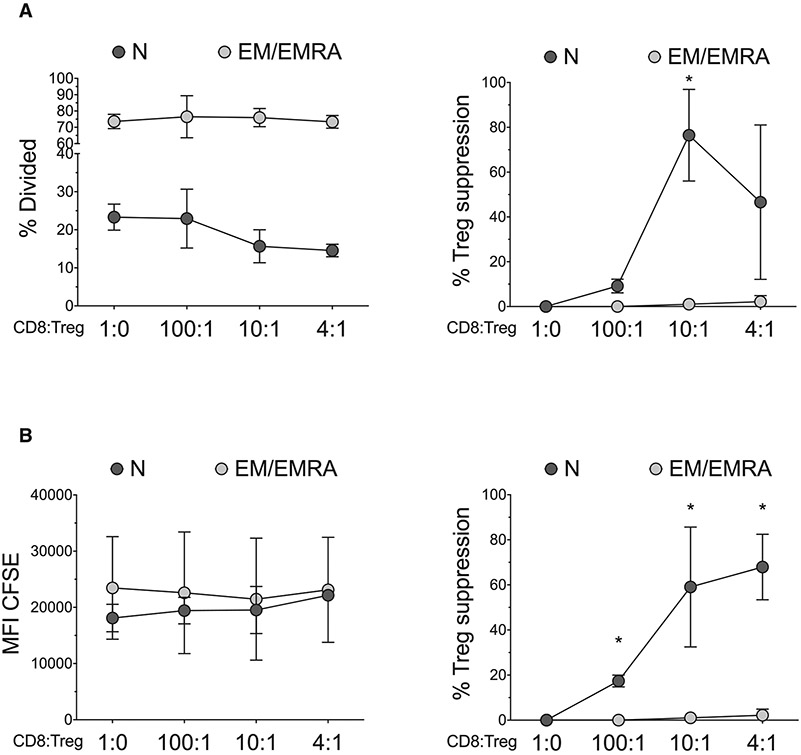
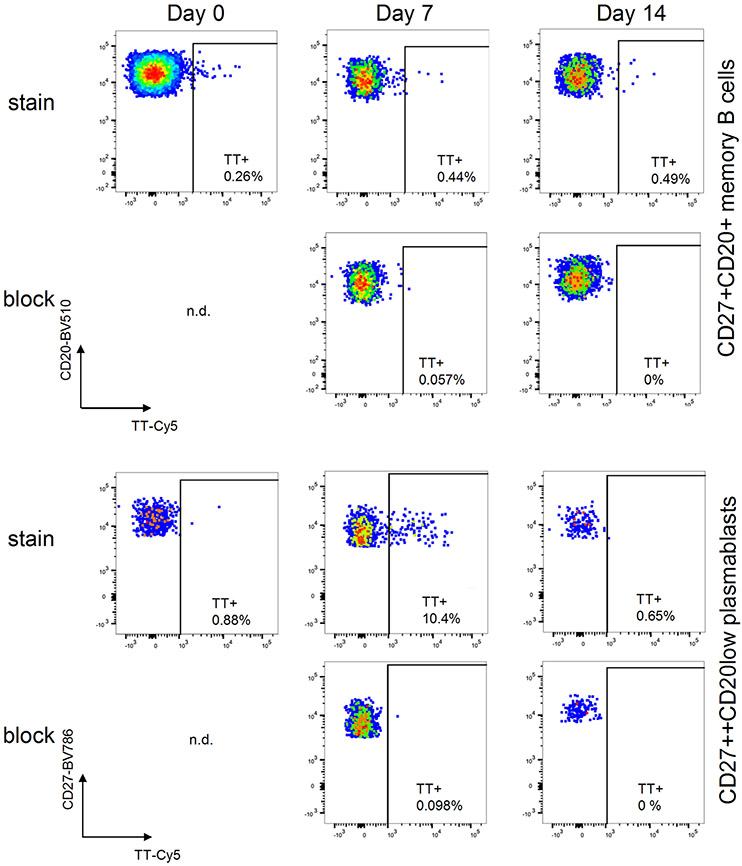
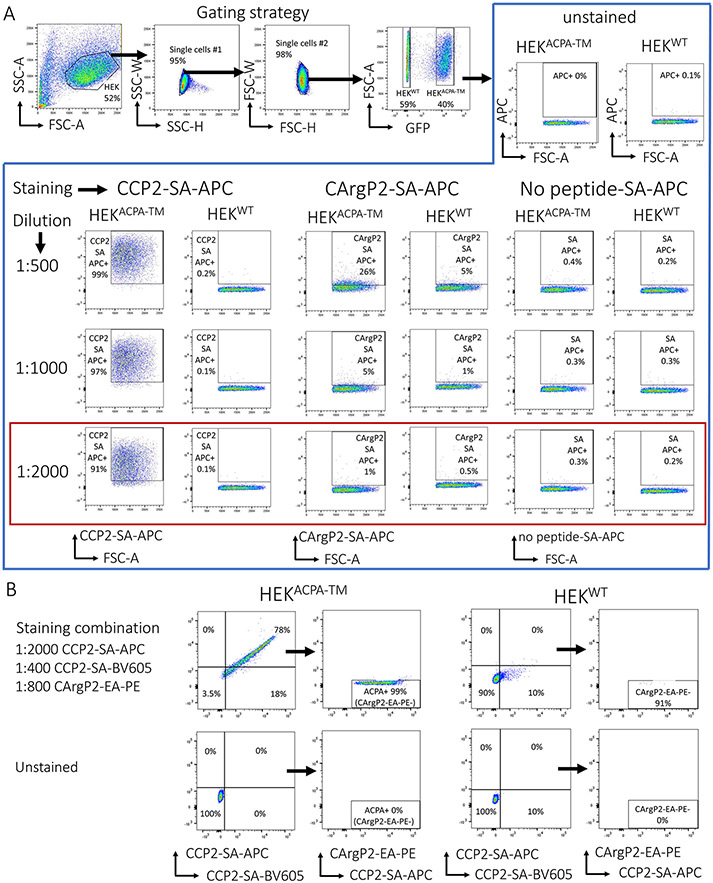
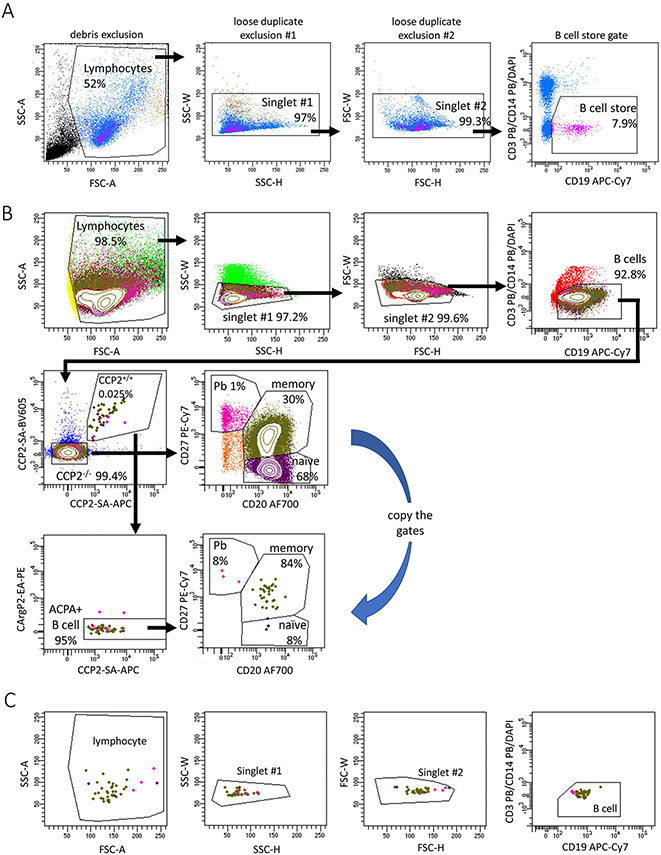
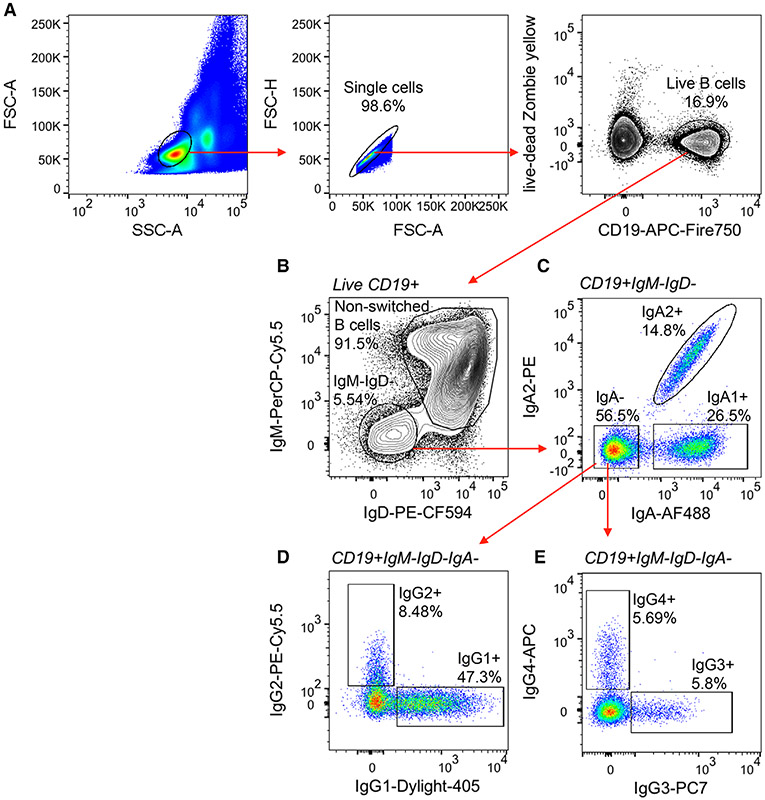
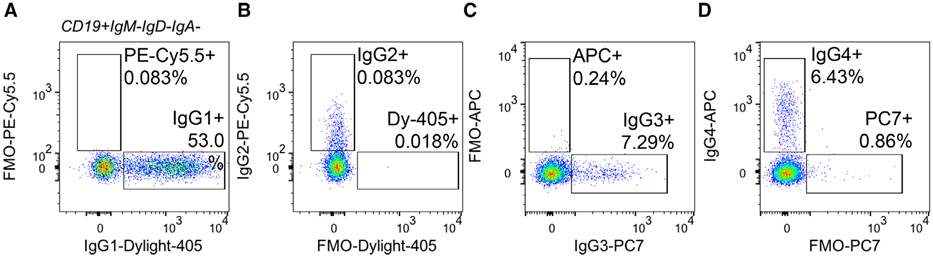
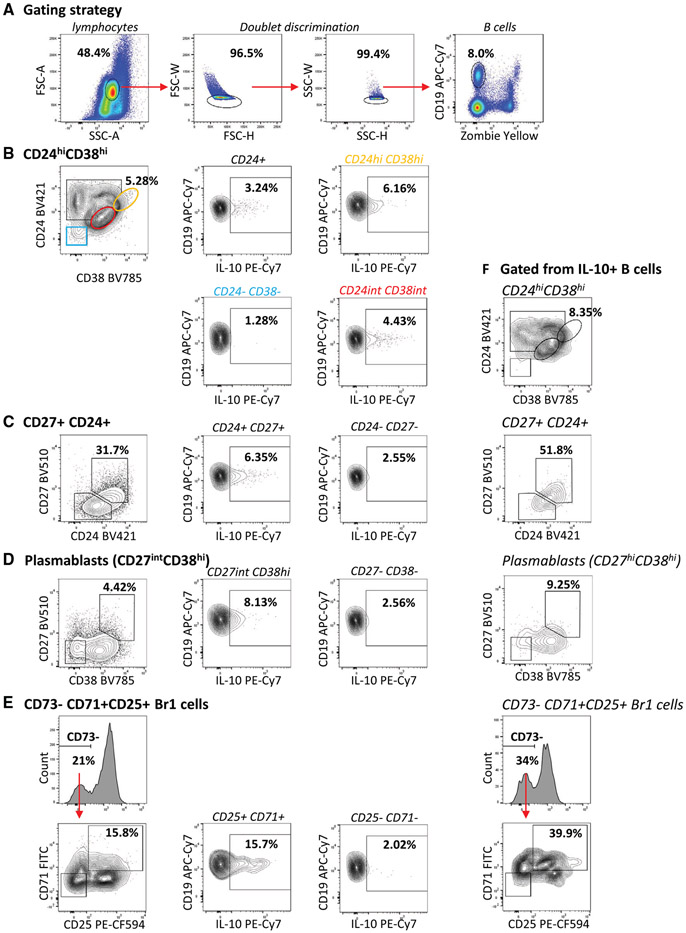
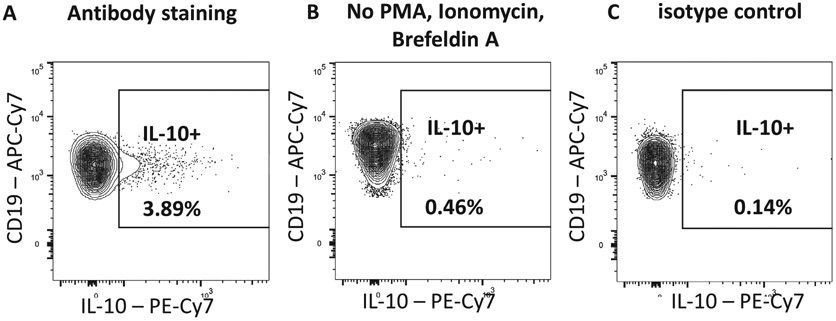
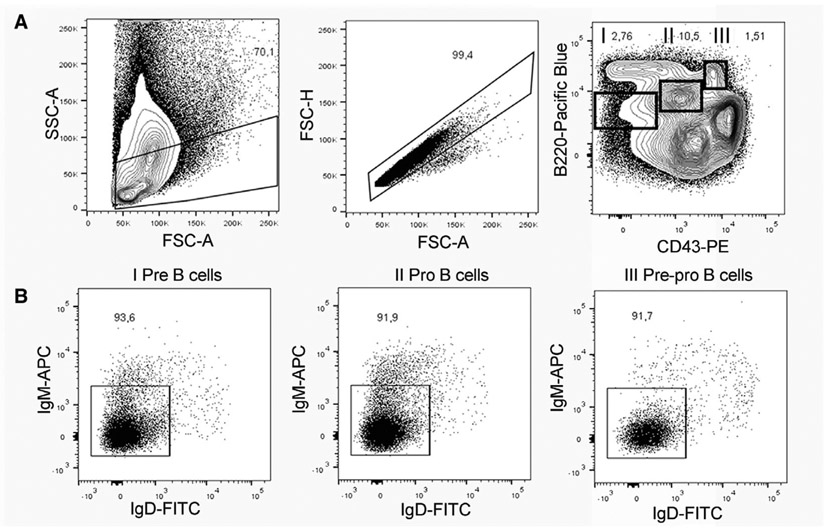
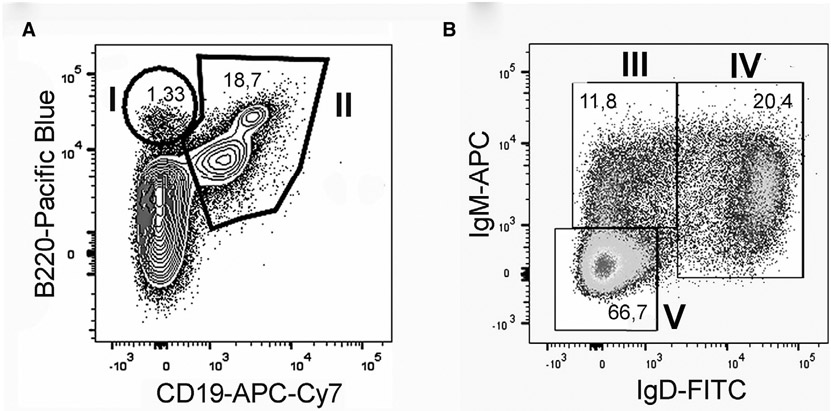
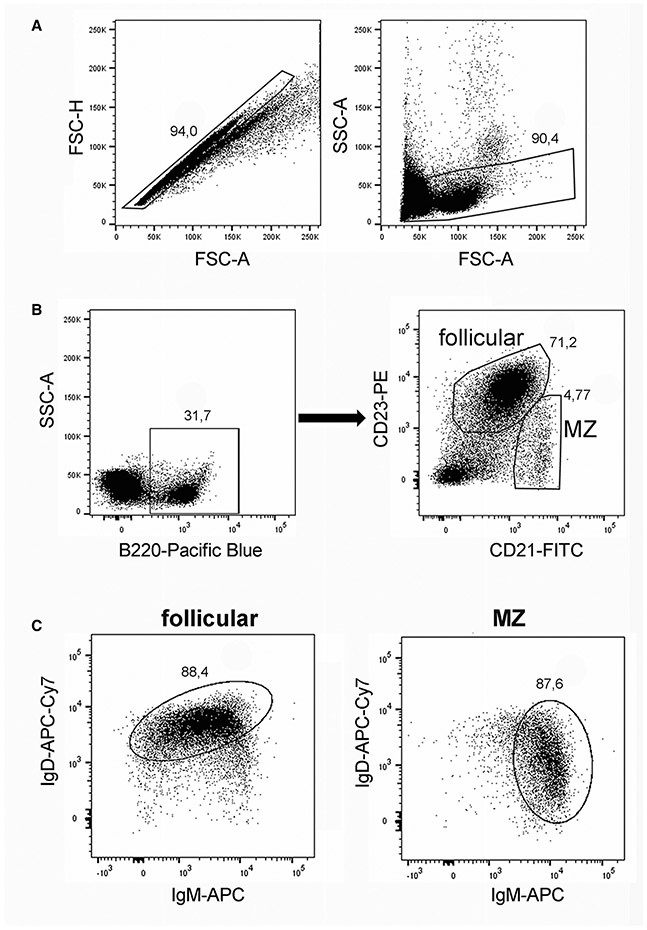

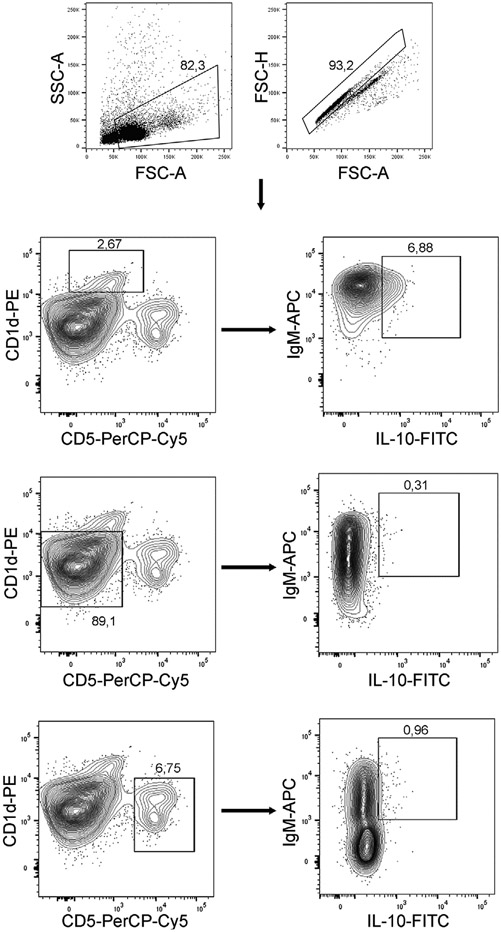
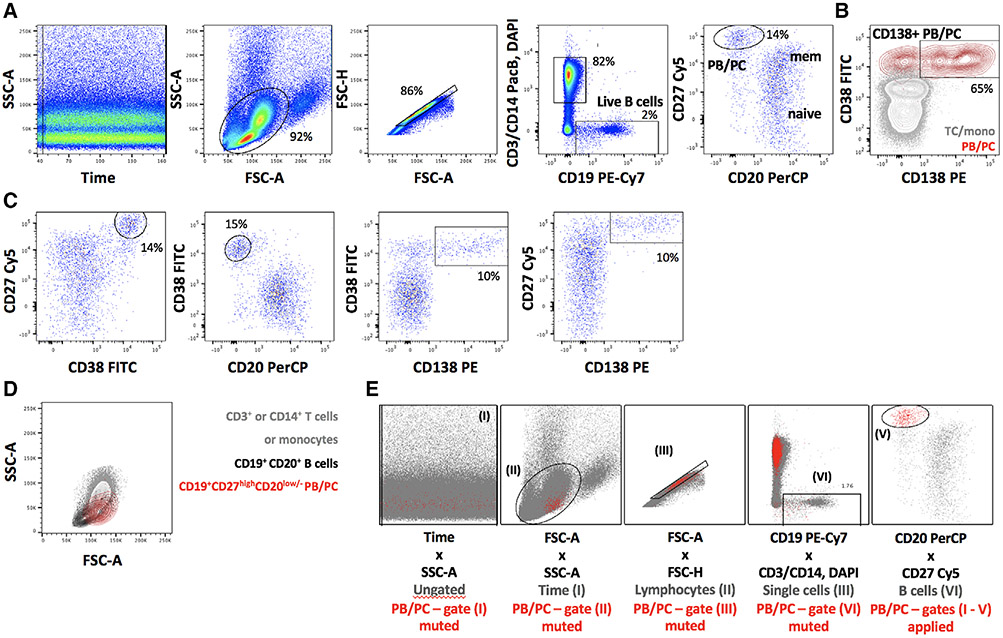
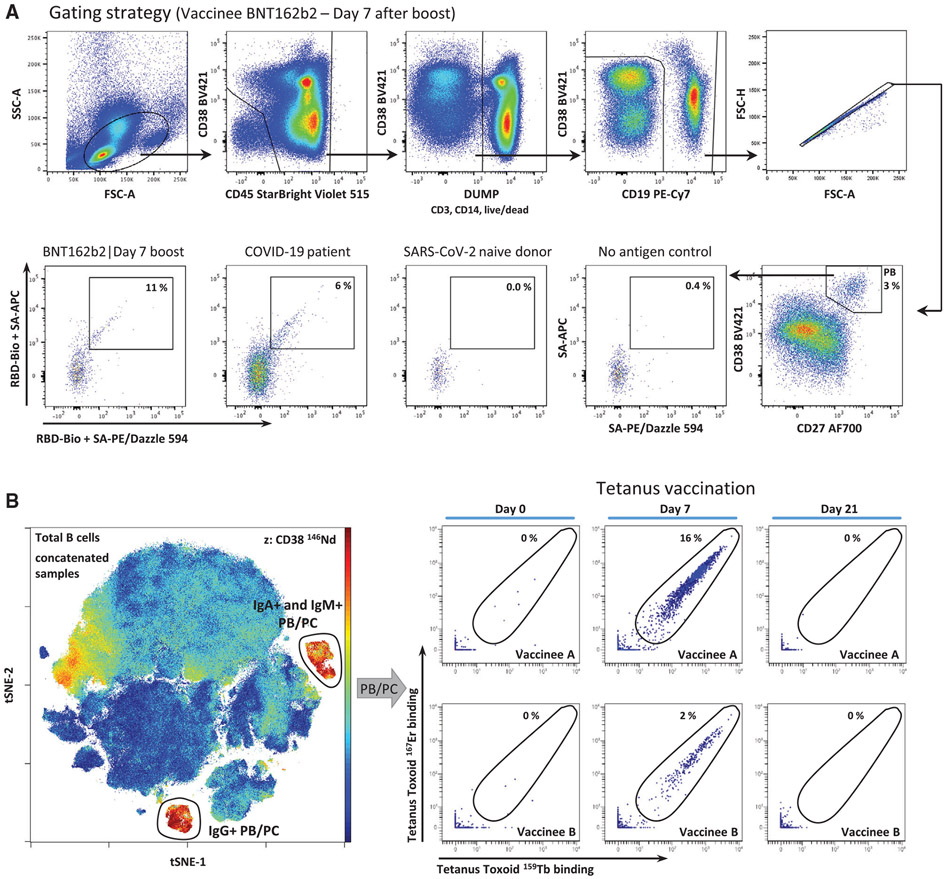
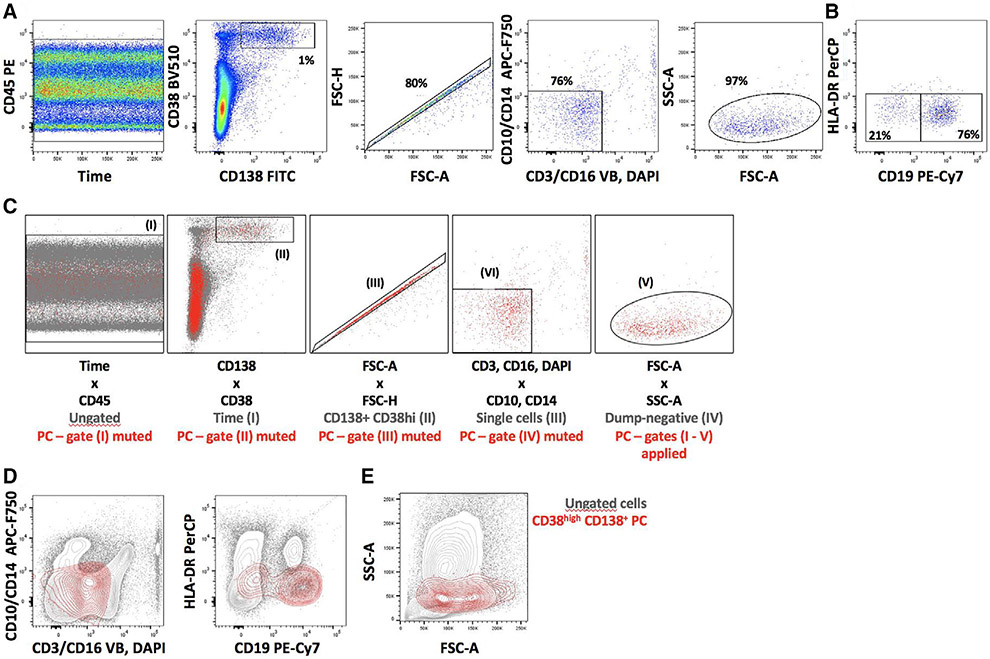
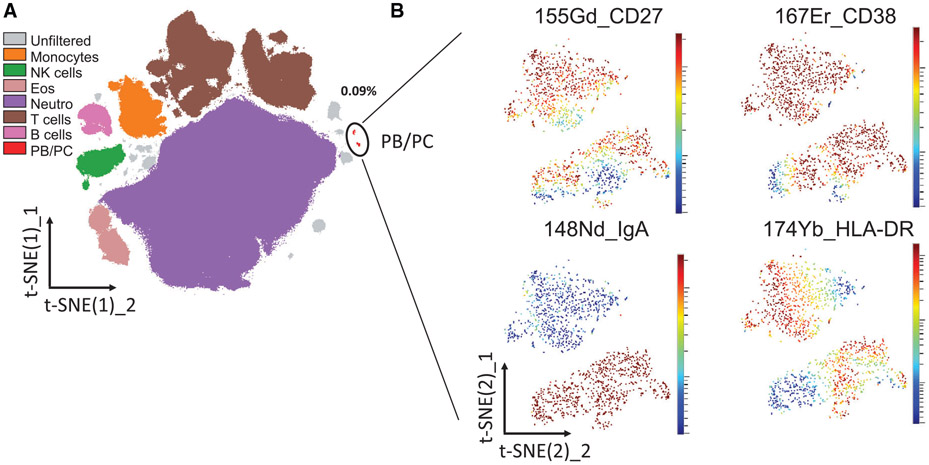
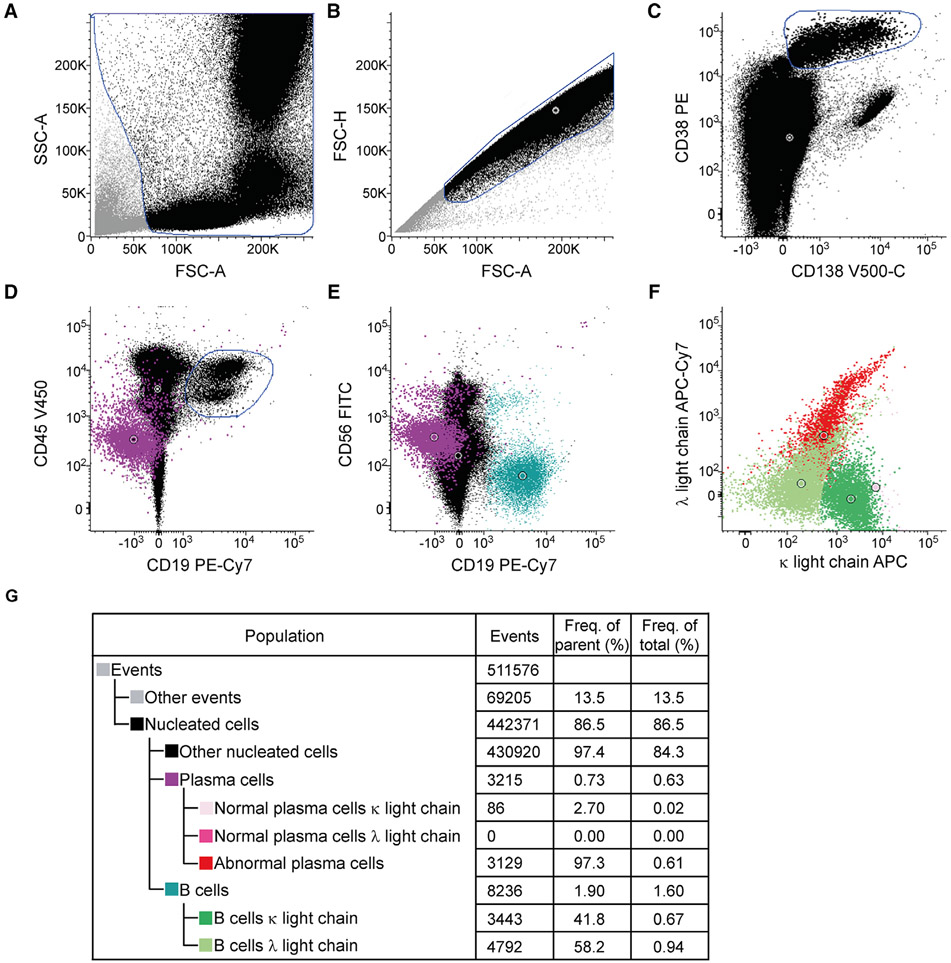
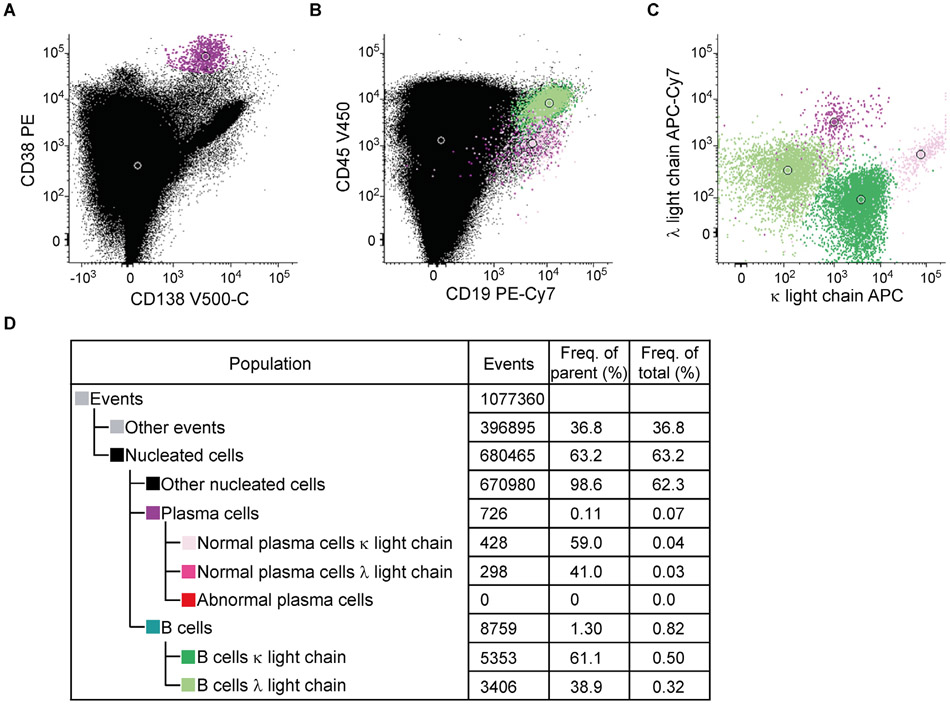
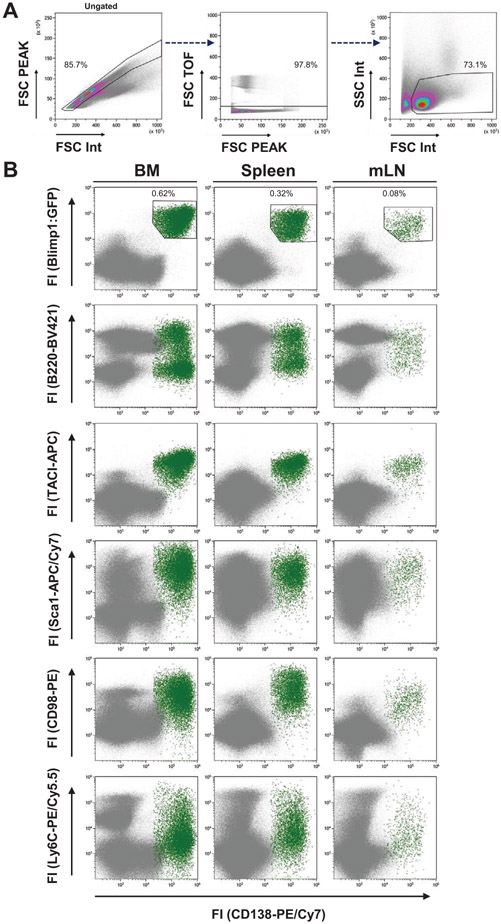
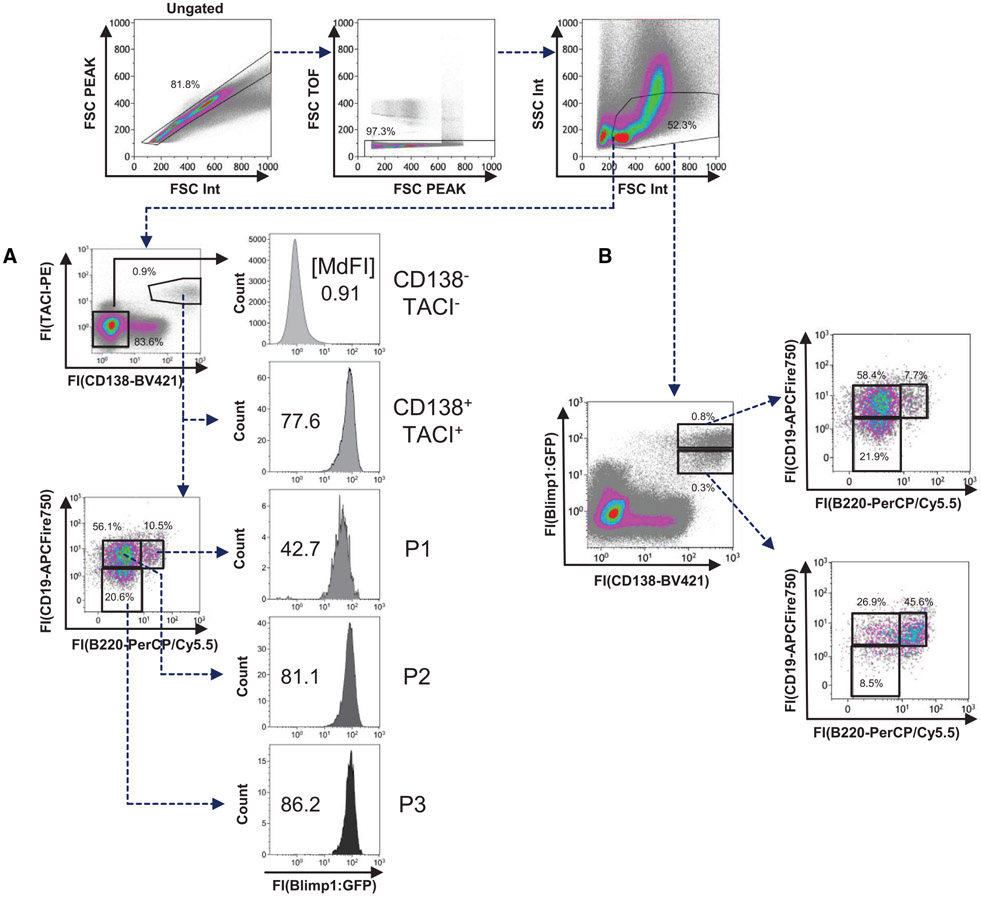
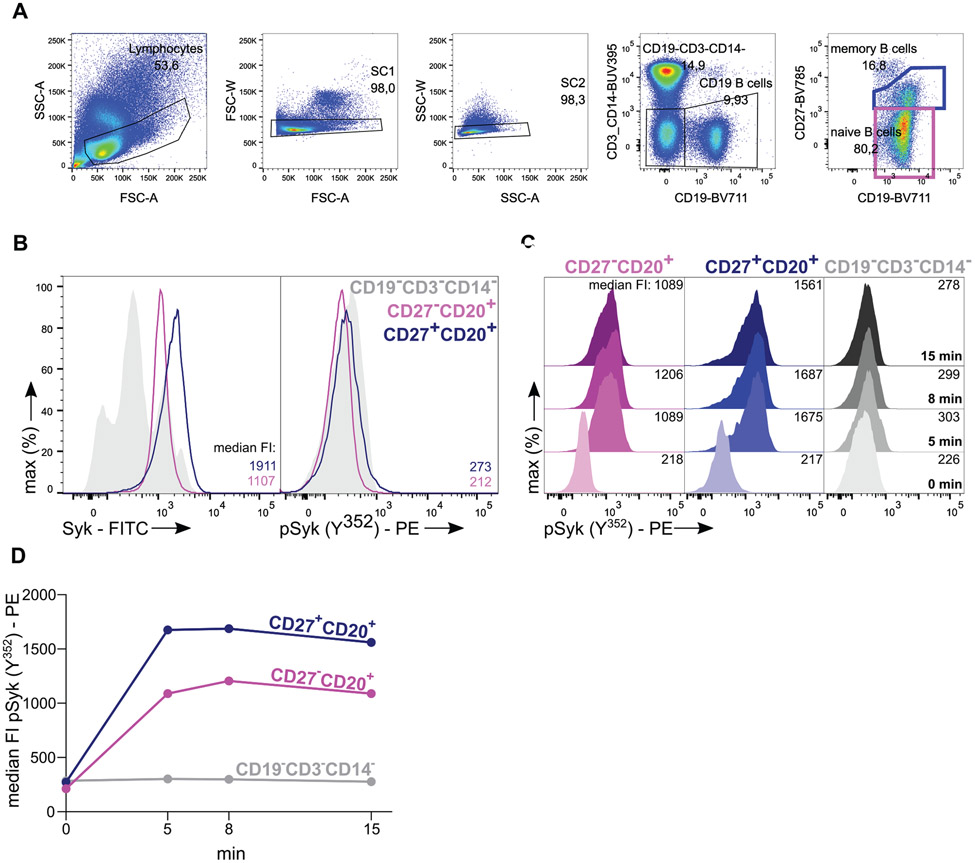
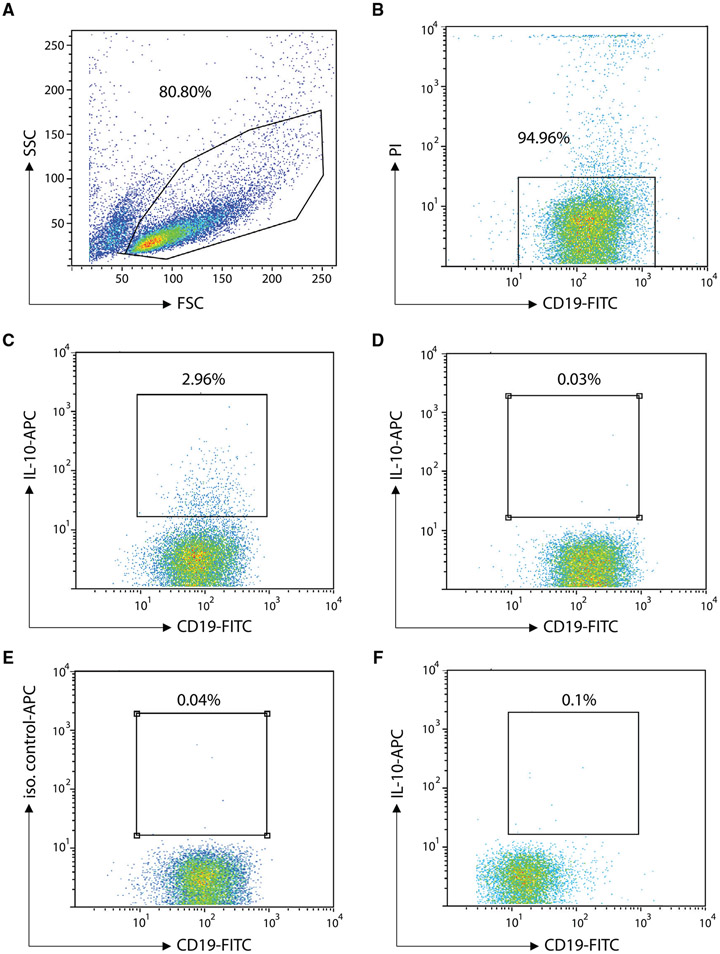
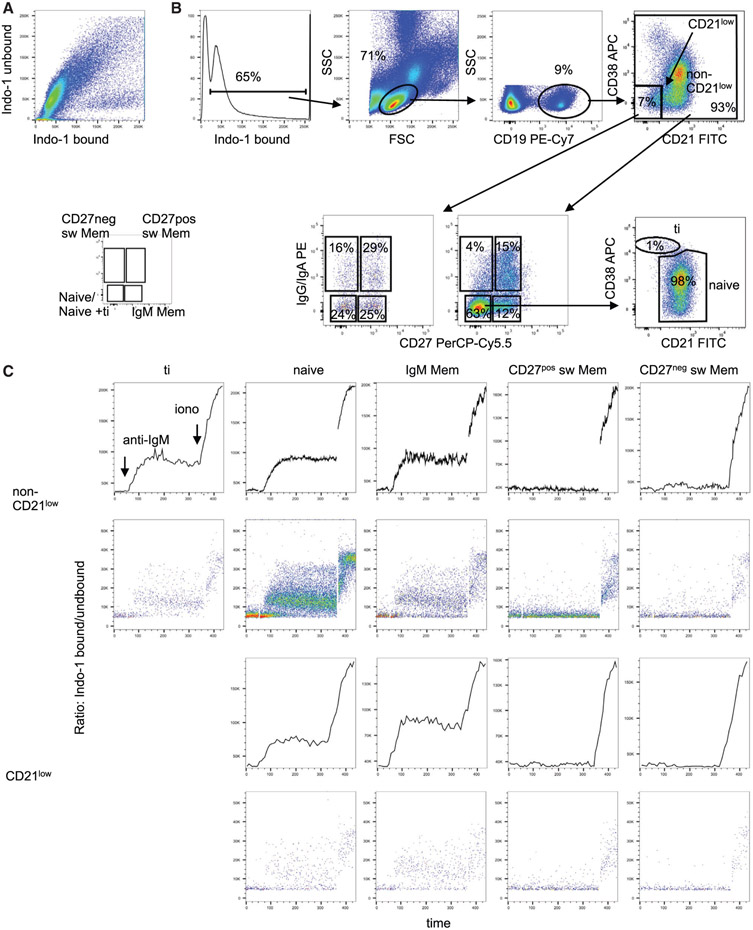
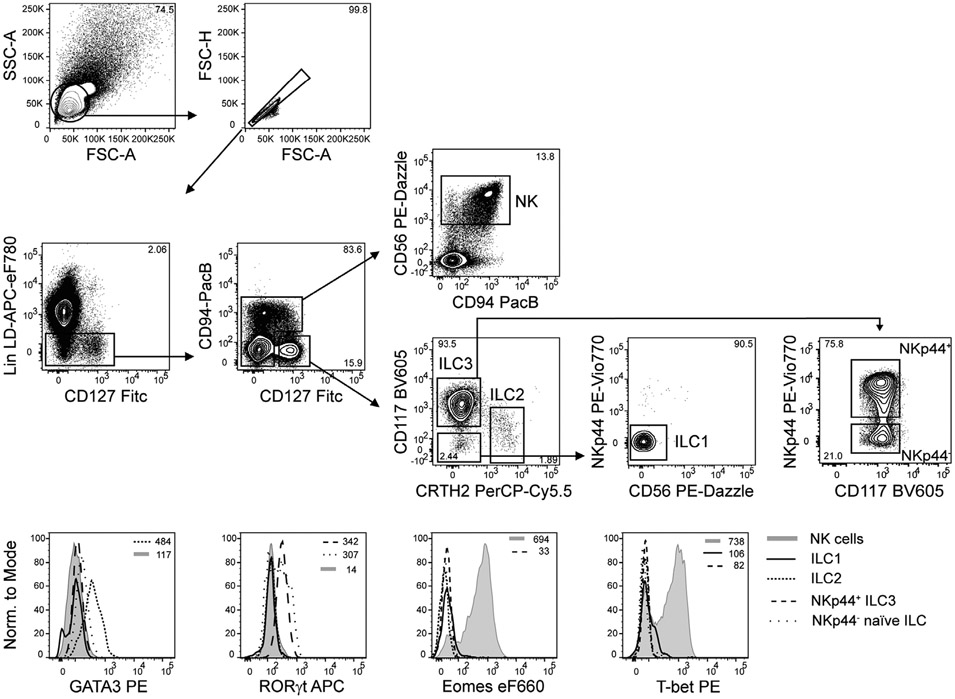
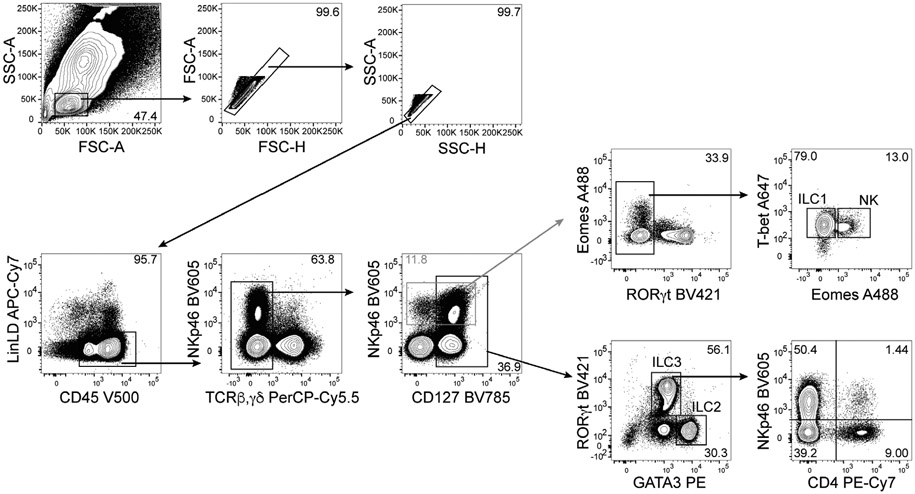
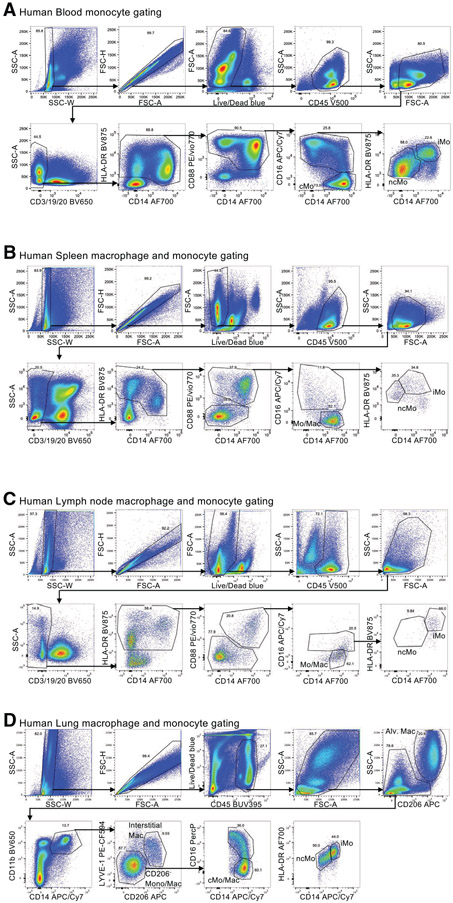
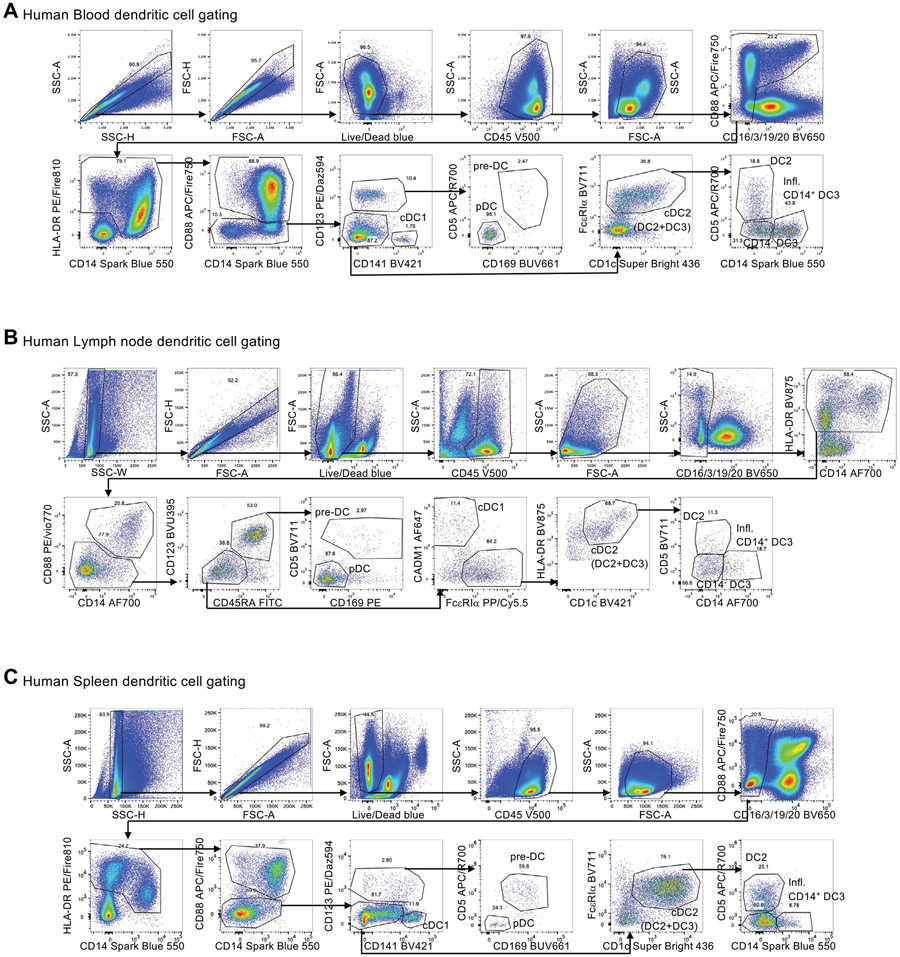

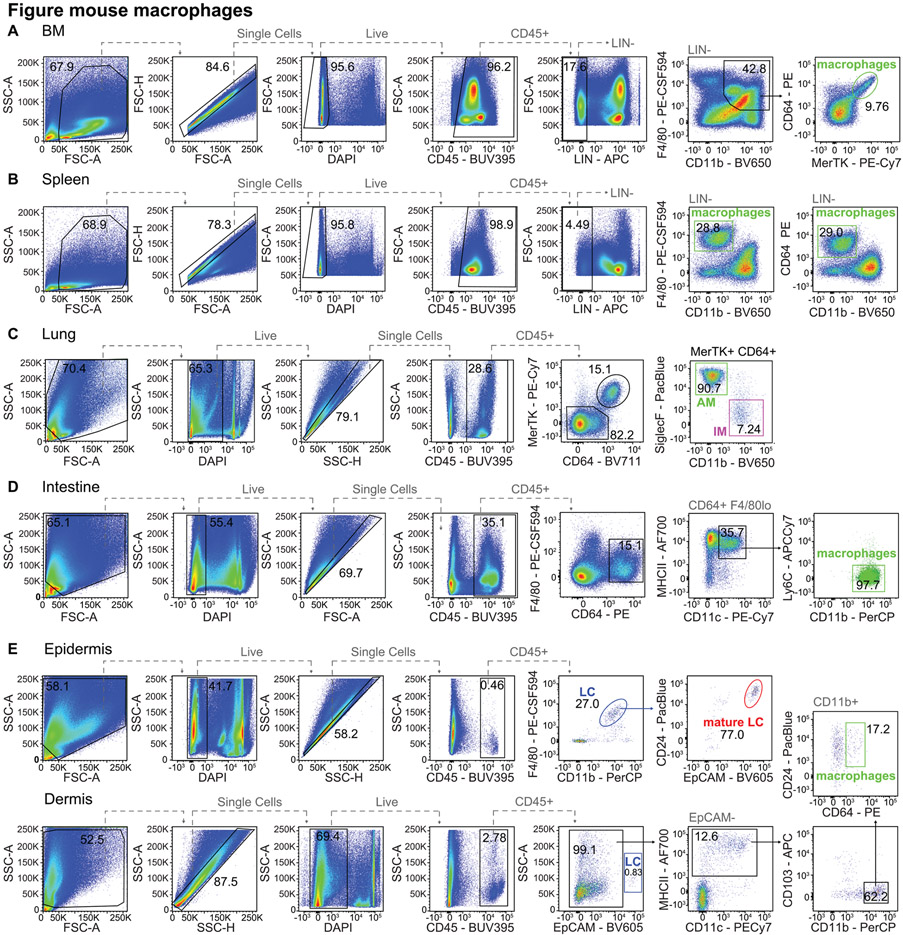
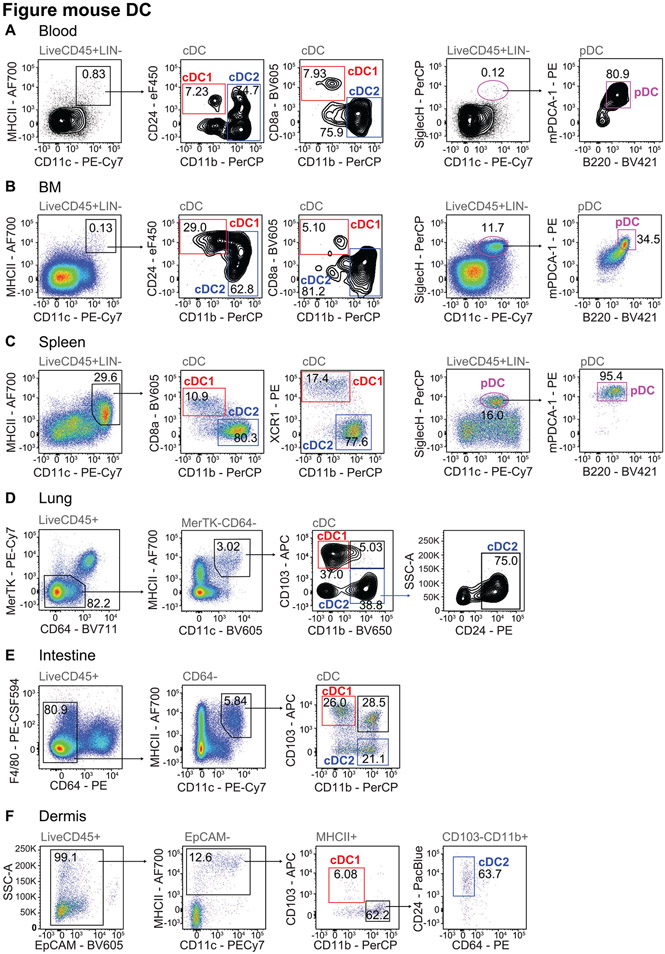
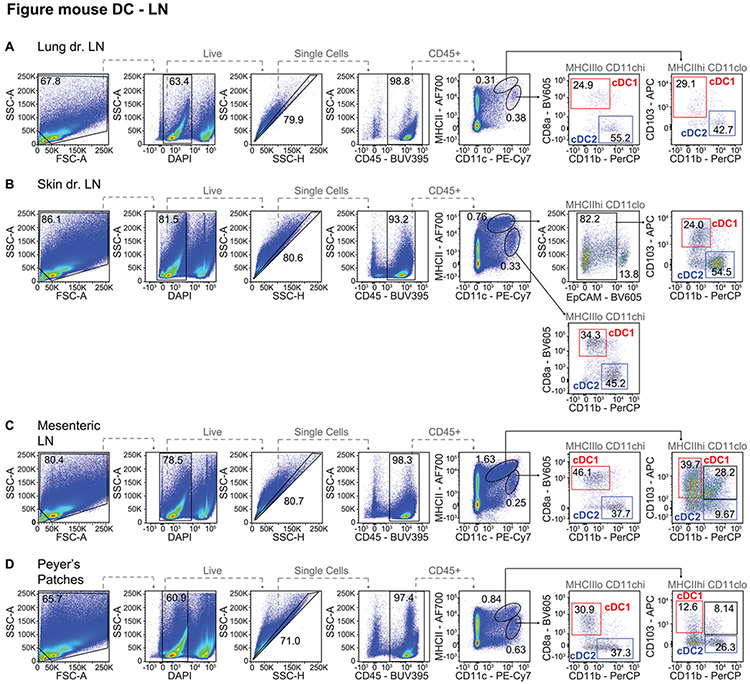
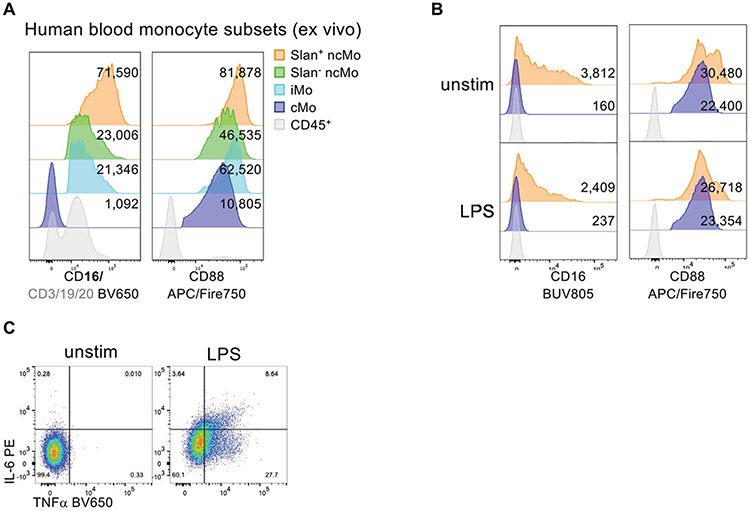
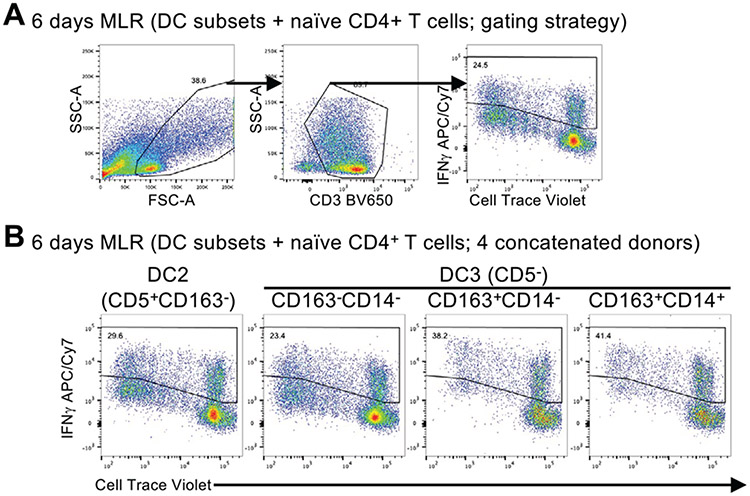
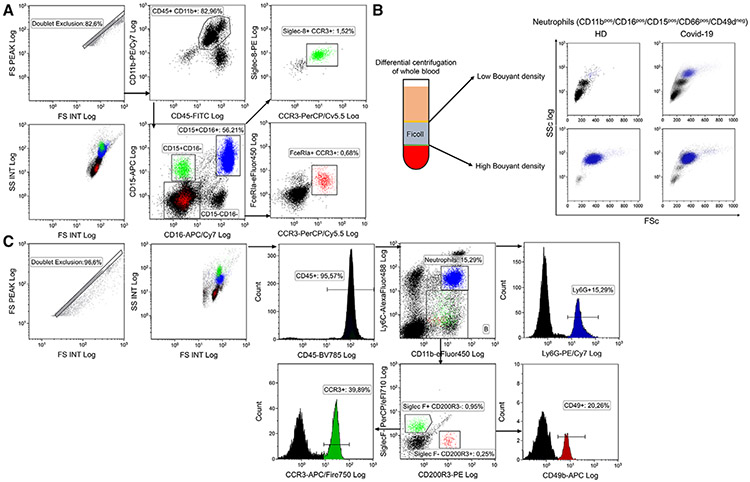
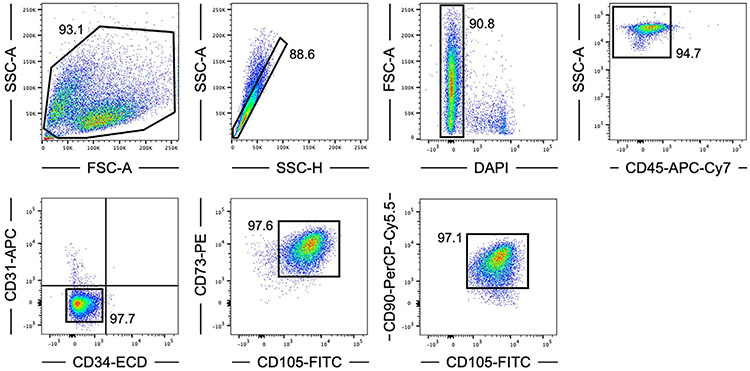
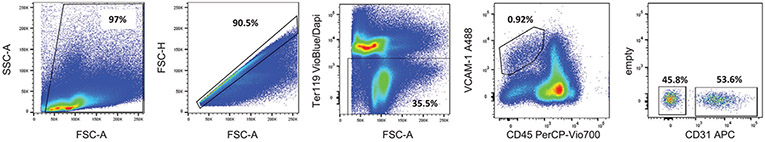
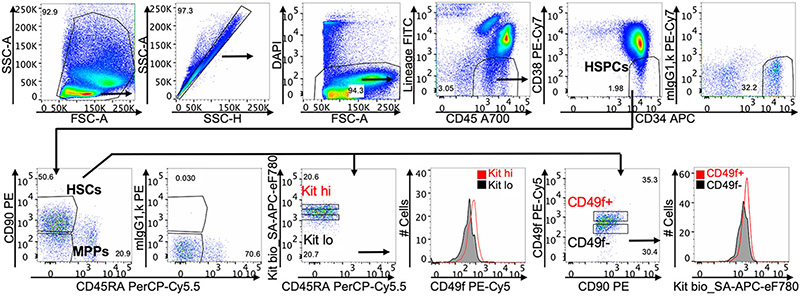
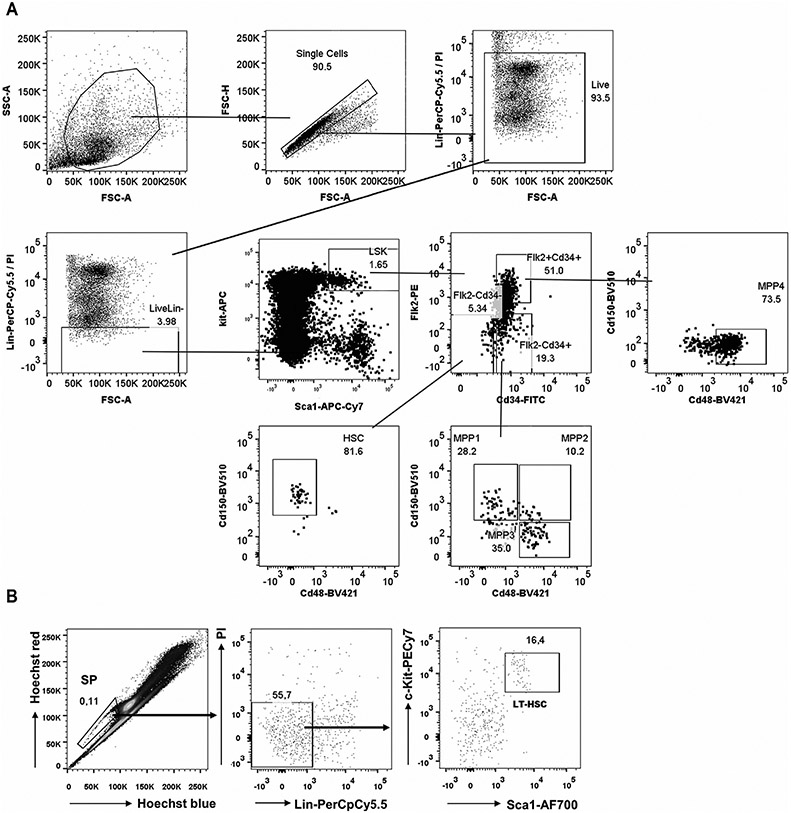
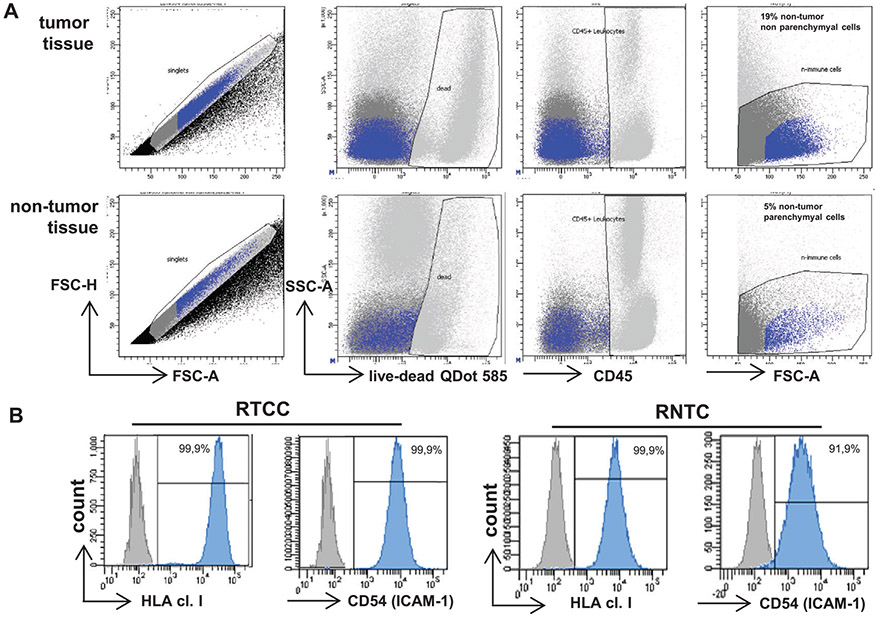
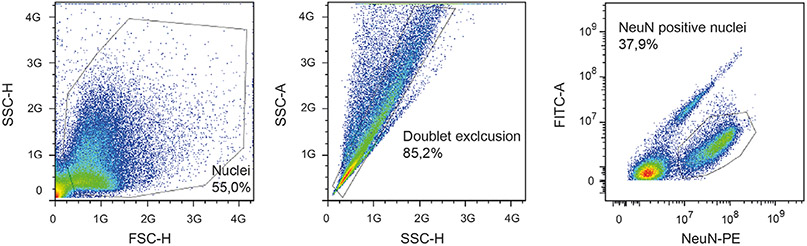
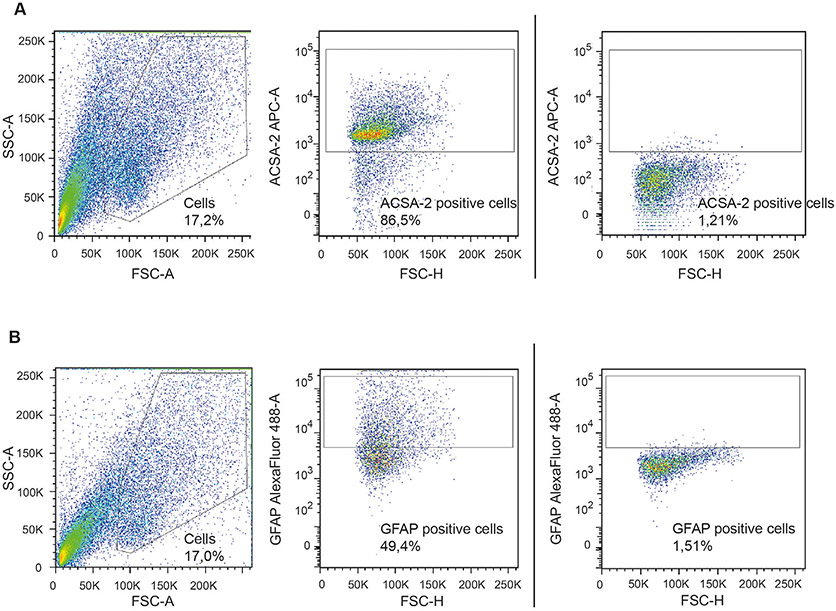
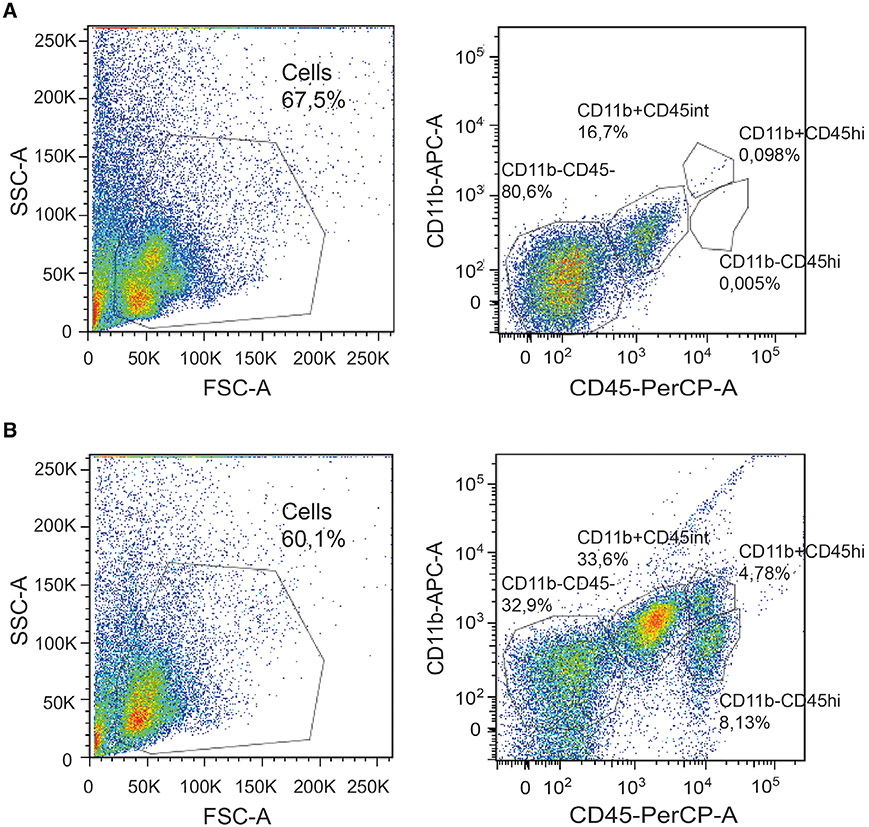
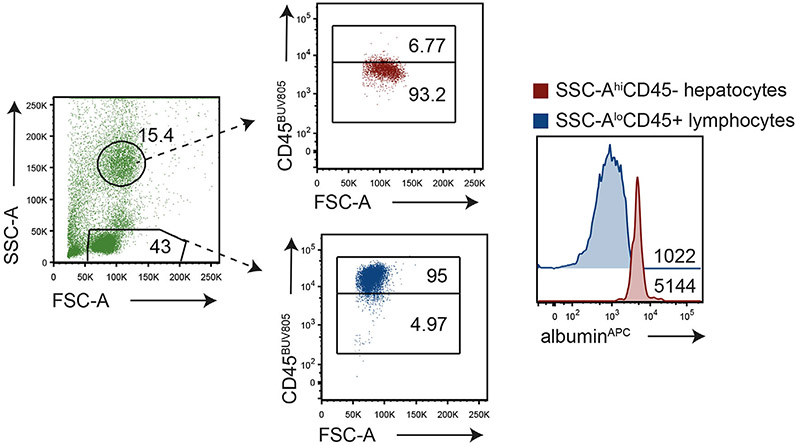
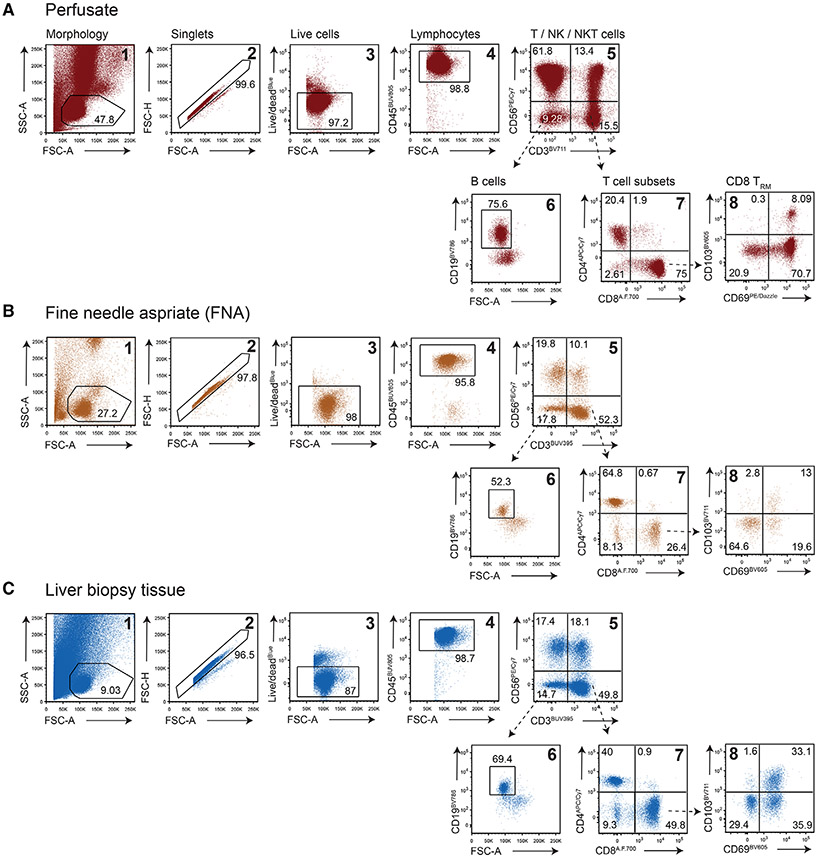
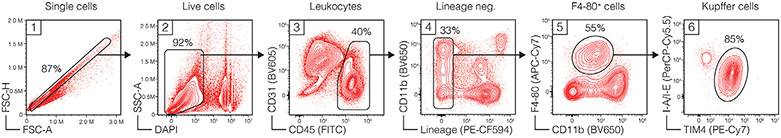
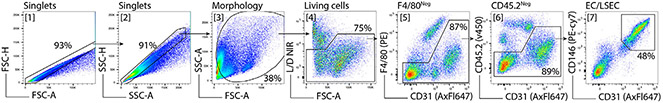
References
-
- Maecker HT and Trotter J, Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry A 2006. 69: 1037–1042. - PubMed
-
- Hulspas R, O’Gorman MR, Wood BL, Gratama JW and Sutherland DR, Considerations for the control of background fluorescence in clinical flow cytometry. Cytom. B. Clin. Cytom 2009. 76: 355–364. - PubMed
-
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay PK and Roederer M, Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat. Protoc 2012. 7: 2067–2079. - PubMed
-
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay P and Roederer M, Quality assurance for polychromatic flow cytometry. Nat. Protoc 2006. 1: 1522–1530. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
