

JNK signaling provides a novel therapeutic target for Rett syndrome
- PMID: 34911542
- PMCID: PMC8675514
- DOI: 10.1186/s12915-021-01190-2
JNK signaling provides a novel therapeutic target for Rett syndrome
Abstract
Background: Rett syndrome (RTT) is a monogenic X-linked neurodevelopmental disorder characterized by loss-of-function mutations in the MECP2 gene, which lead to structural and functional changes in synapse communication, and impairments of neural activity at the basis of cognitive deficits that progress from an early age. While the restoration of MECP2 in animal models has been shown to rescue some RTT symptoms, gene therapy intervention presents potential side effects, and with gene- and RNA-editing approaches still far from clinical application, strategies focusing on signaling pathways downstream of MeCP2 may provide alternatives for the development of more effective therapies in vivo. Here, we investigate the role of the c-Jun N-terminal kinase (JNK) stress pathway in the pathogenesis of RTT using different animal and cell models and evaluate JNK inhibition as a potential therapeutic approach.
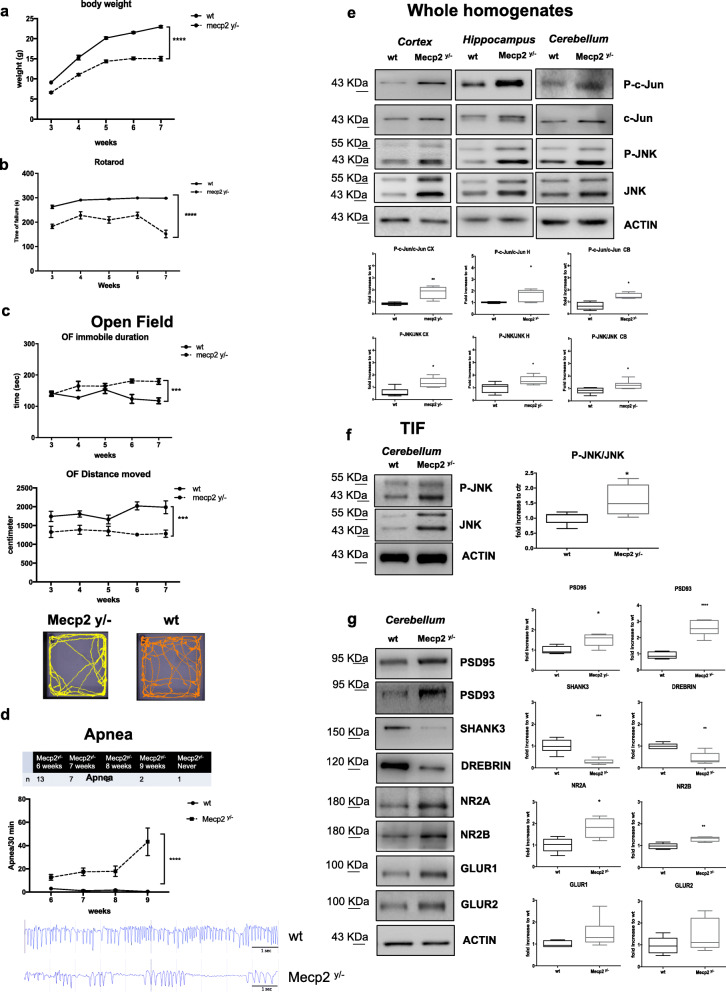
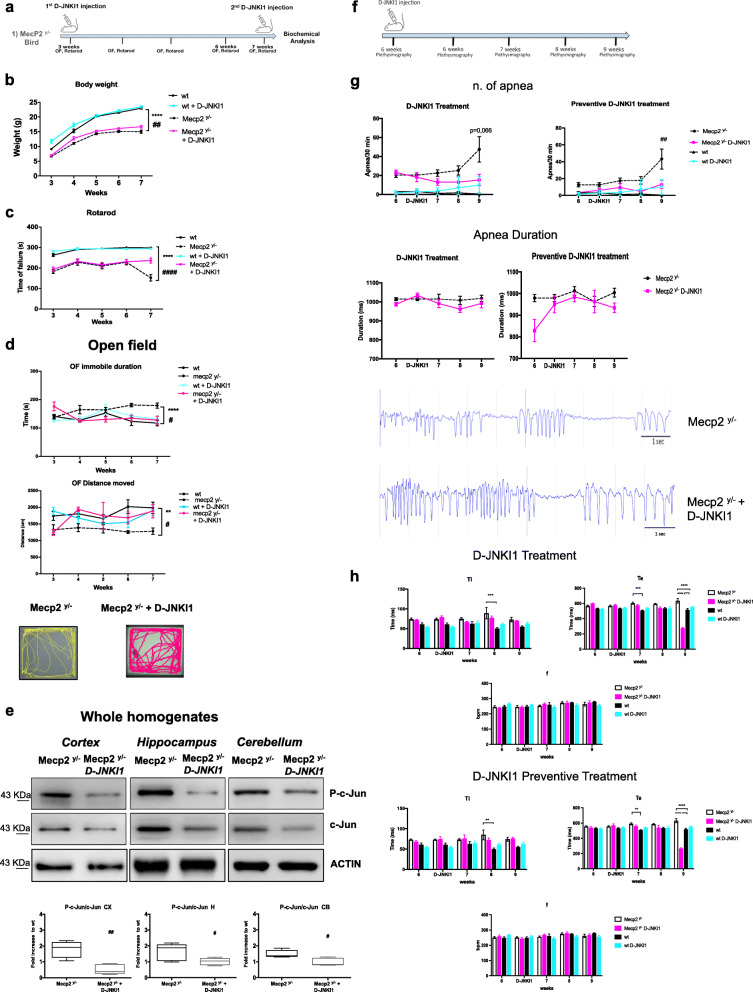
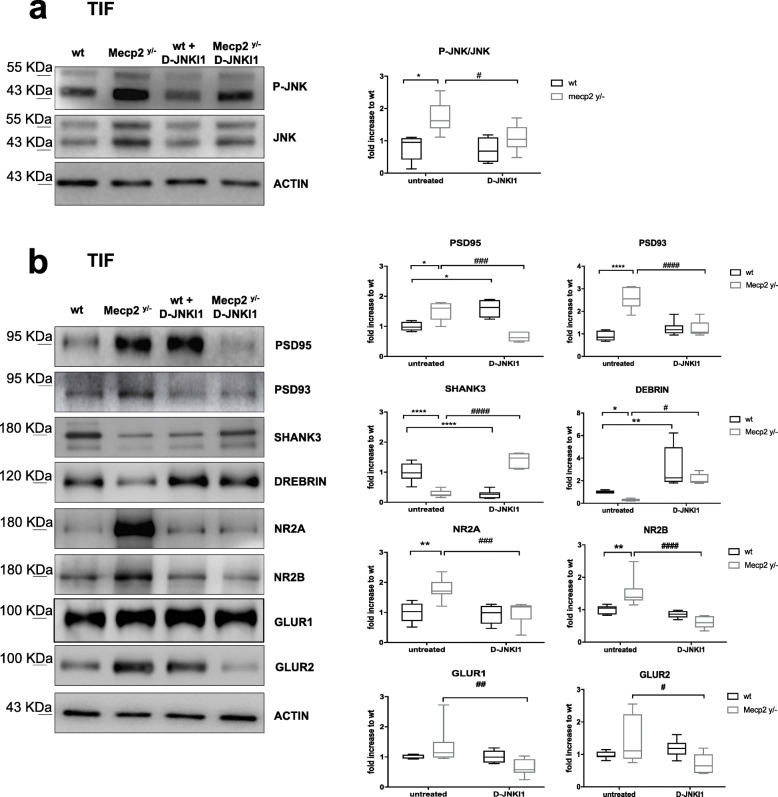
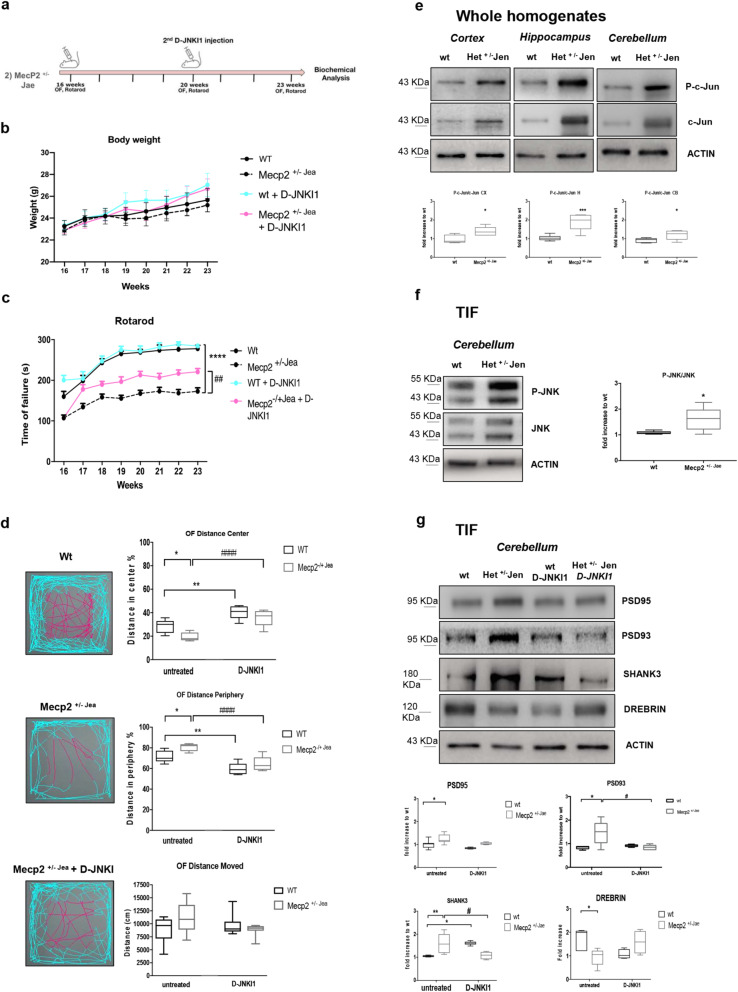
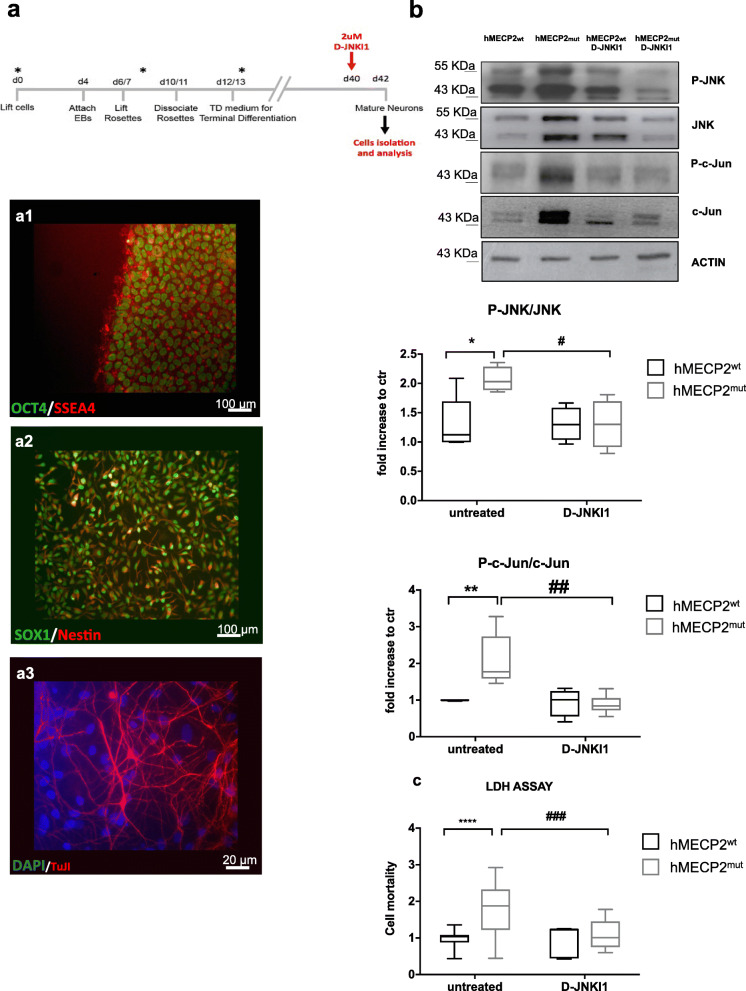
Results: We discovered that the c-Jun N-terminal kinase (JNK) stress pathway is activated in Mecp2-knockout, Mecp2-heterozygous mice, and in human MECP2-mutated iPSC neurons. The specific JNK inhibitor, D-JNKI1, promotes recovery of body weight and locomotor impairments in two mouse models of RTT and rescues their dendritic spine alterations. Mecp2-knockout presents intermittent crises of apnea/hypopnea, one of the most invalidating RTT pathological symptoms, and D-JNKI1 powerfully reduces this breathing dysfunction. Importantly, we discovered that also neurons derived from hiPSC-MECP2 mut show JNK activation, high-phosphorylated c-Jun levels, and cell death, which is not observed in the isogenic control wt allele hiPSCs. Treatment with D-JNKI1 inhibits neuronal death induced by MECP2 mutation in hiPSCs mut neurons.
Conclusions: As a summary, we found altered JNK signaling in models of RTT and suggest that D-JNKI1 treatment prevents clinical symptoms, with coherent results at the cellular, molecular, and functional levels. This is the first proof of concept that JNK plays a key role in RTT and its specific inhibition offers a new and potential therapeutic tool to tackle RTT.
Keywords: Apnea; D-JNKI1; MECP2; Neurodevelopmental disease; Neuroprotection; Synaptic dysfunction.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Percy AK. Rett syndrome: Current status and new vistas. Neurol Clin. 2002;20:1125–1141. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Jørgensen HF, Bird A. MeCP2 and other methyl-CpG binding proteins. Ment Retard Dev Disabil Res Rev. 2002;8:87–93. - PubMed
-
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. - PubMed
-
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
