

Ontogeny and Vulnerabilities of Drug-Tolerant Persisters in HER2+ Breast Cancer
- PMID: 34911733
- PMCID: PMC8983469
- DOI: 10.1158/2159-8290.CD-20-1265
Ontogeny and Vulnerabilities of Drug-Tolerant Persisters in HER2+ Breast Cancer
Abstract
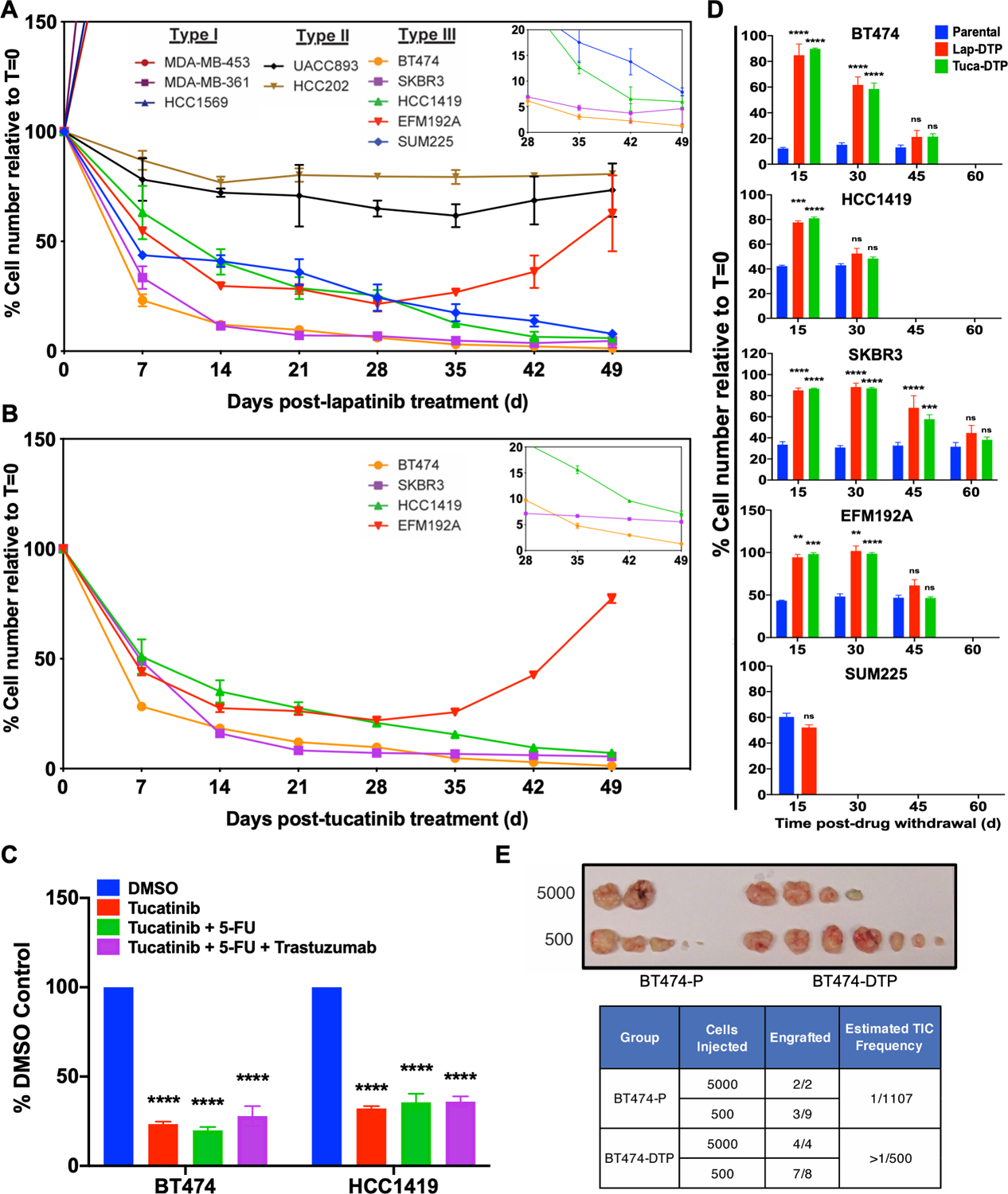
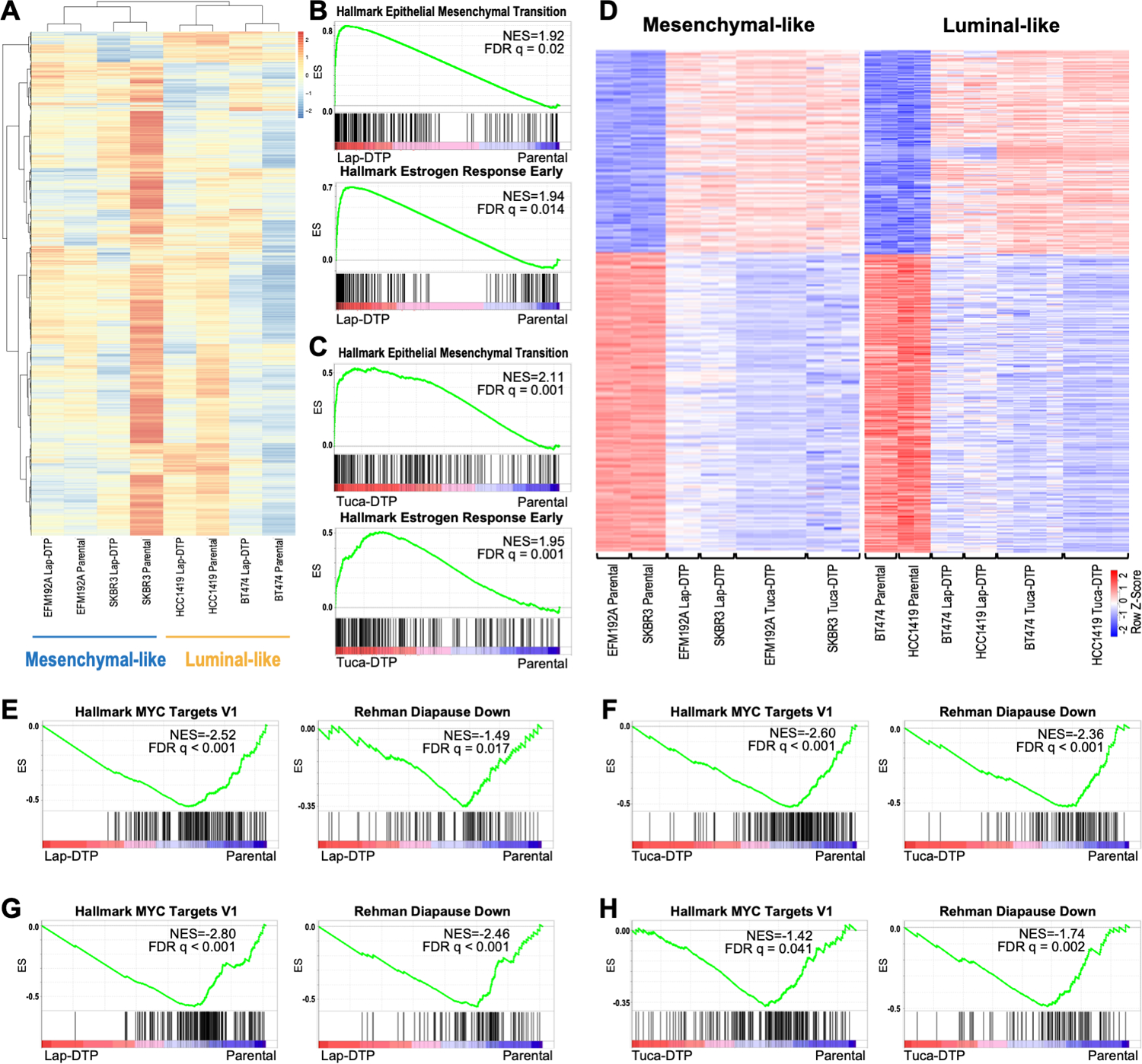
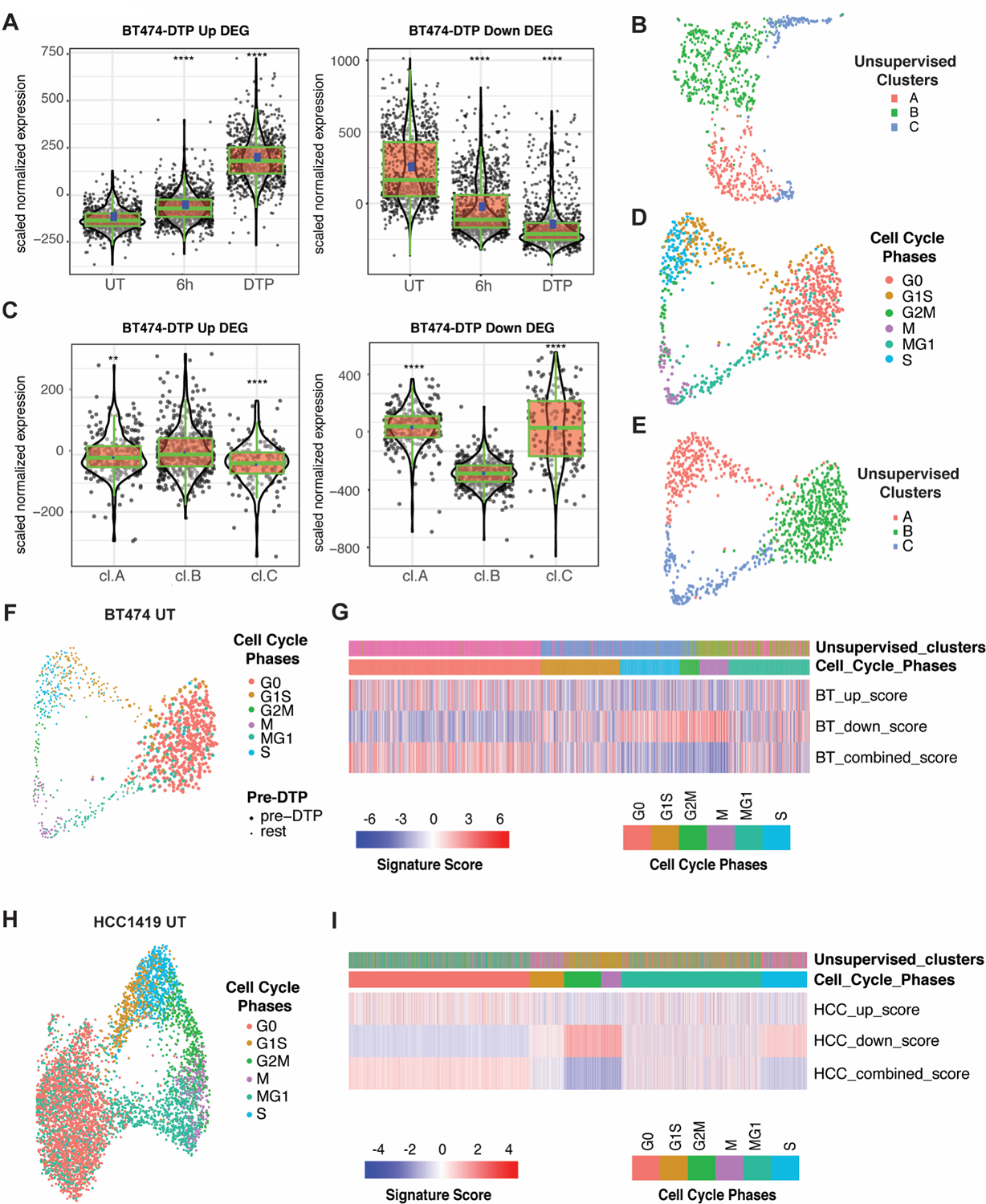
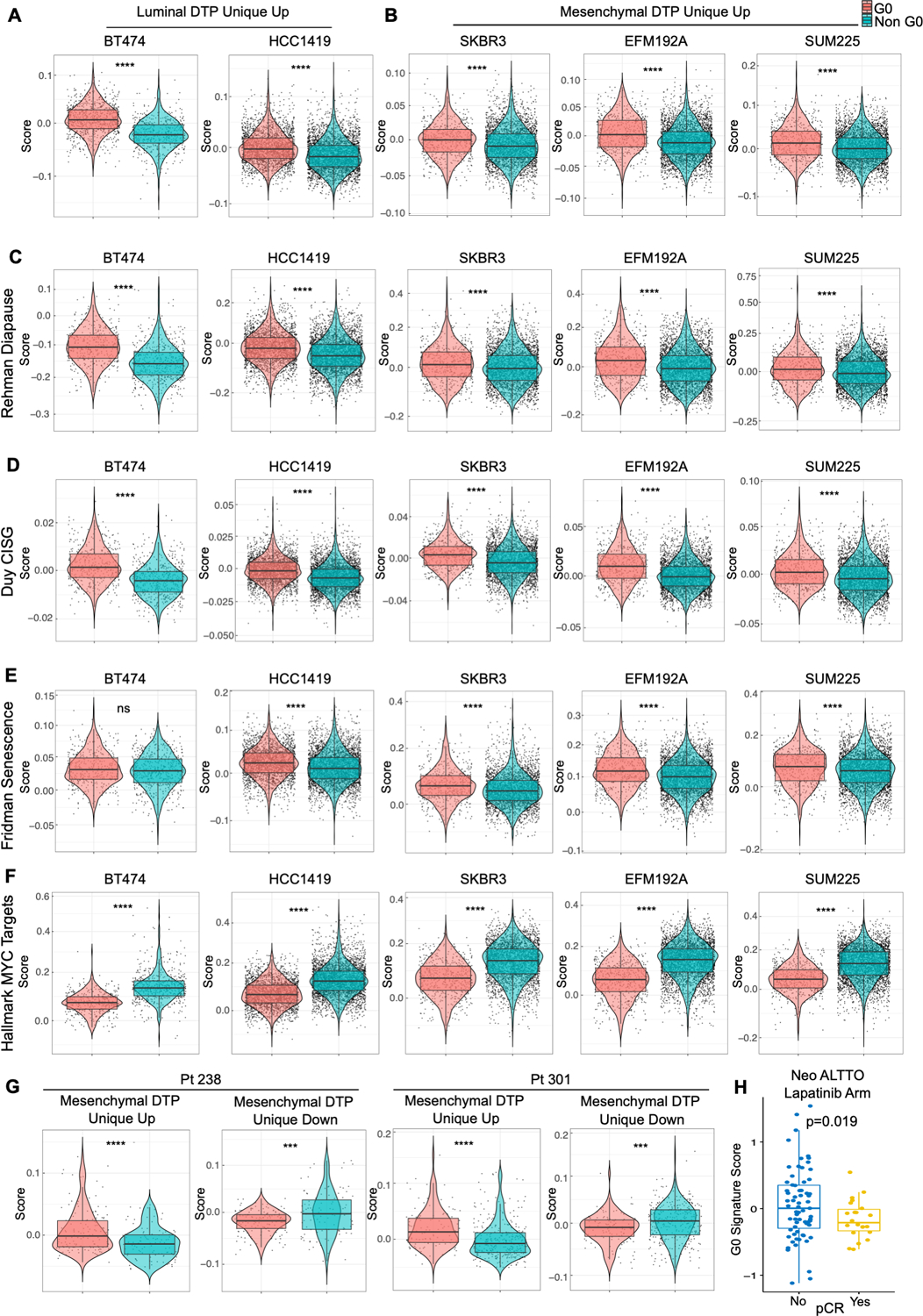
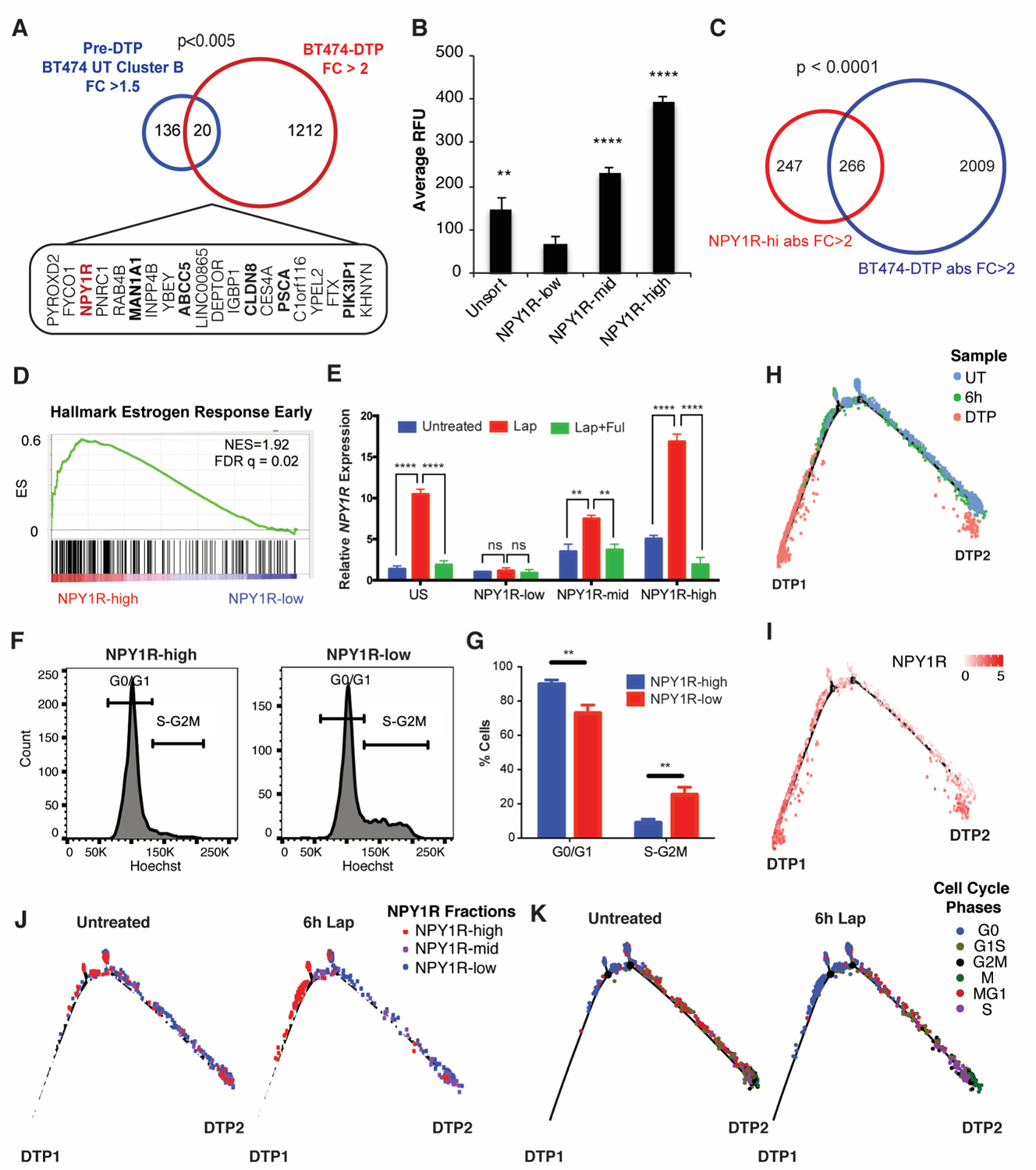
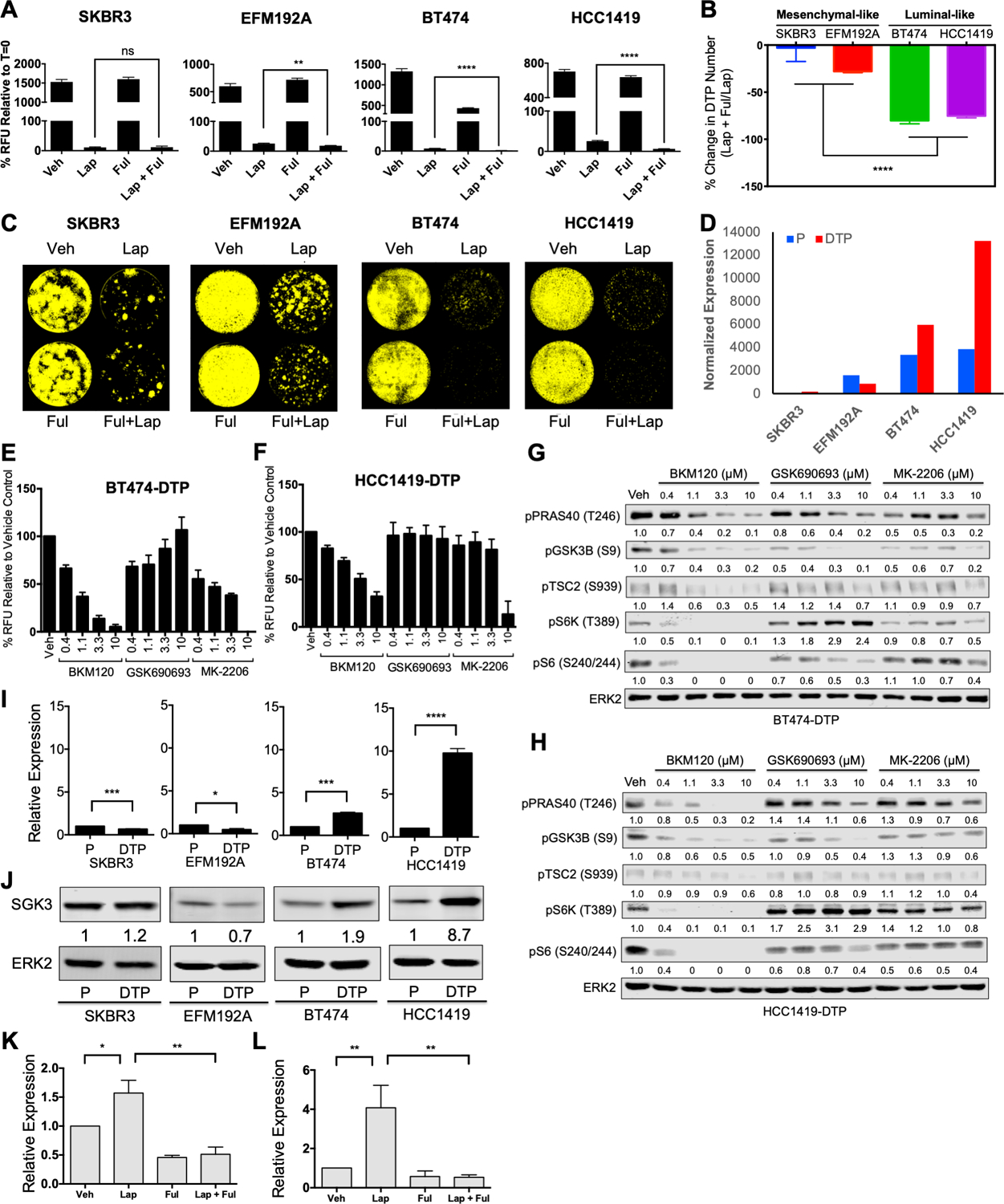
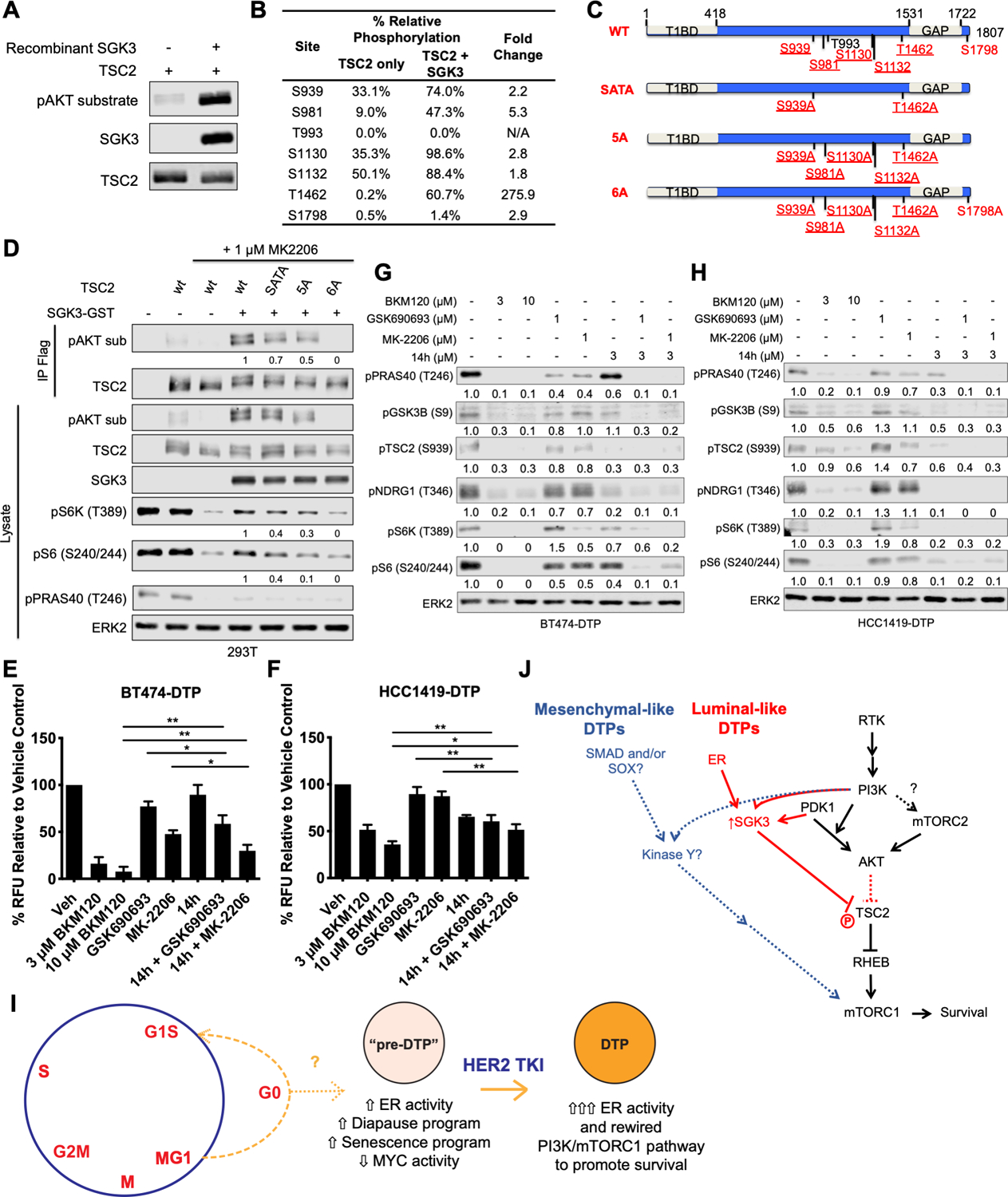
Resistance to targeted therapies is an important clinical problem in HER2-positive (HER2+) breast cancer. "Drug-tolerant persisters" (DTP), a subpopulation of cancer cells that survive via reversible, nongenetic mechanisms, are implicated in resistance to tyrosine kinase inhibitors (TKI) in other malignancies, but DTPs following HER2 TKI exposure have not been well characterized. We found that HER2 TKIs evoke DTPs with a luminal-like or a mesenchymal-like transcriptome. Lentiviral barcoding/single-cell RNA sequencing reveals that HER2+ breast cancer cells cycle stochastically through a "pre-DTP" state, characterized by a G0-like expression signature and enriched for diapause and/or senescence genes. Trajectory analysis/cell sorting shows that pre-DTPs preferentially yield DTPs upon HER2 TKI exposure. Cells with similar transcriptomes are present in HER2+ breast tumors and are associated with poor TKI response. Finally, biochemical experiments indicate that luminal-like DTPs survive via estrogen receptor-dependent induction of SGK3, leading to rewiring of the PI3K/AKT/mTORC1 pathway to enable AKT-independent mTORC1 activation.
Significance: DTPs are implicated in resistance to anticancer therapies, but their ontogeny and vulnerabilities remain unclear. We find that HER2 TKI-DTPs emerge from stochastically arising primed cells ("pre-DTPs") that engage either of two distinct transcriptional programs upon TKI exposure. Our results provide new insights into DTP ontogeny and potential therapeutic vulnerabilities. This article is highlighted in the In This Issue feature, p. 873.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Pondé N, Brandão M, El-Hachem G, Werbrouck E, Piccart M. Treatment of advanced HER2-positive breast cancer: 2018 and beyond. Cancer Treat Rev 2018;67:10–20. - PubMed
-
- Scaltriti M, Rojo F, Ocaña A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst 2007;99:628–38. - PubMed
-
- Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther 2001;1:85–94. - PubMed
-
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006;355:2733–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
